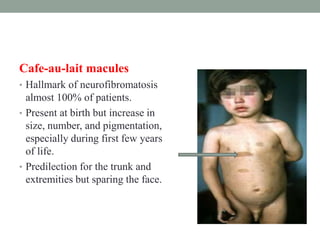
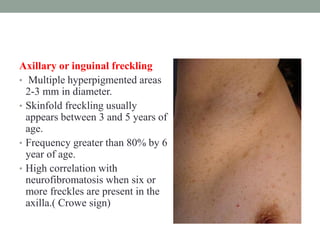
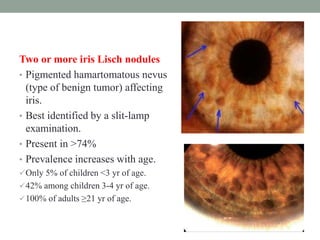
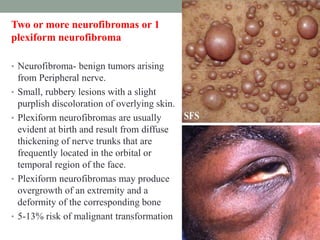
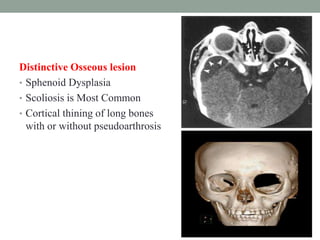
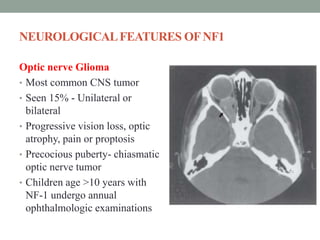
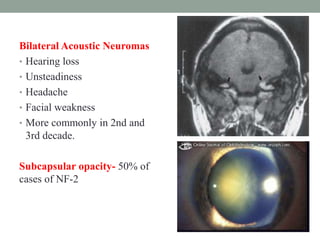
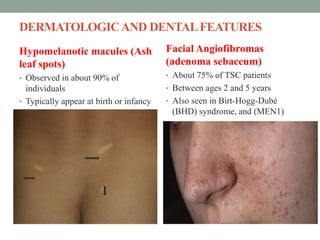
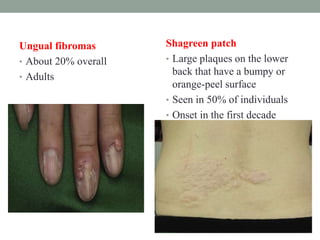
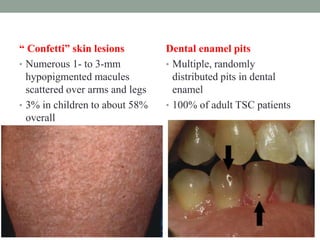
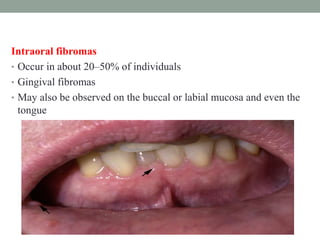
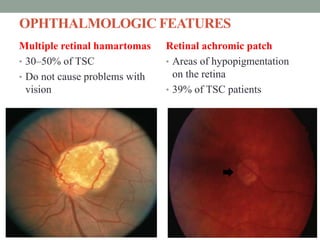
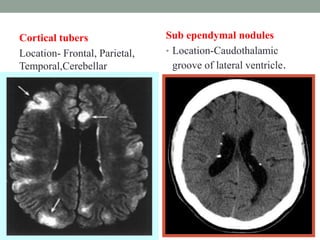
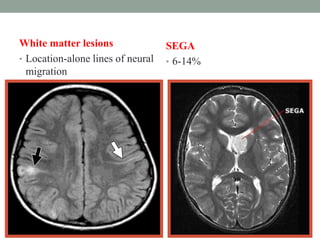
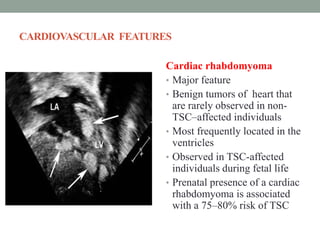
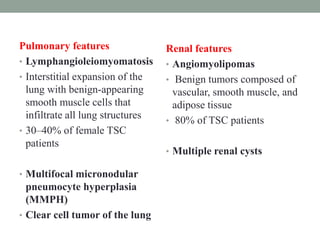
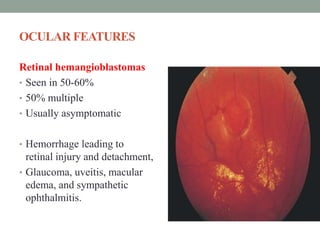
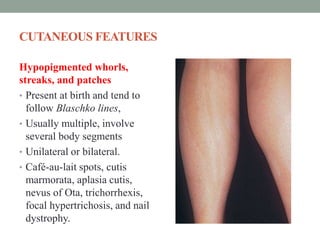
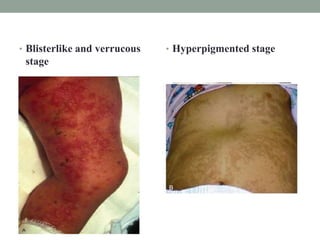
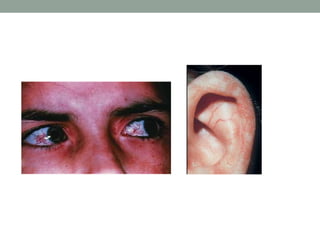
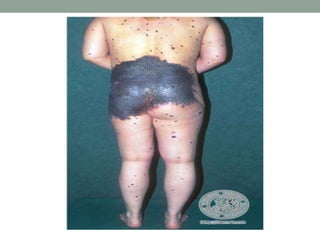
This document discusses neurocutaneous syndromes, which are disorders characterized by abnormalities of the skin and central nervous system. Some key syndromes mentioned include neurofibromatosis, tuberous sclerosis, Sturge-Weber syndrome, and Von Hippel-Lindau syndrome. Neurofibromatosis type 1 is described in detail, outlining its diagnostic criteria involving cafe-au-lait spots, freckling, and tumors. Tuberous sclerosis is also summarized, noting its diagnostic criteria involve tumors in multiple organ systems. Sturge-Weber syndrome links a port-wine stain on the face with leptomeningeal angiomas in the brain.





































































































































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)

































