Downloaded 449 times








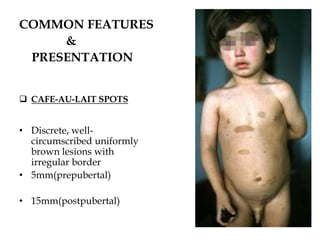
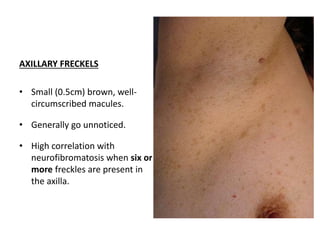
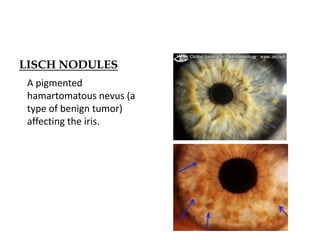
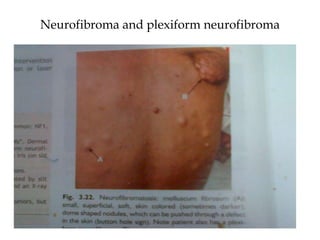


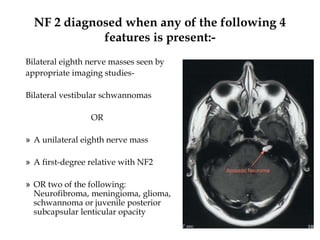

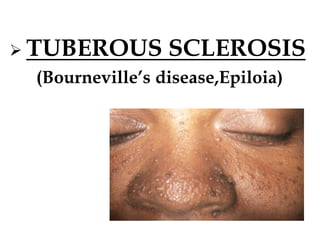

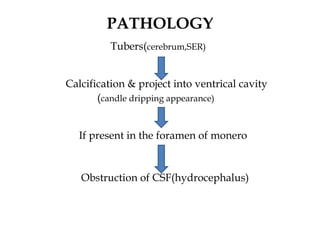
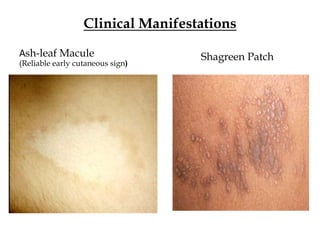
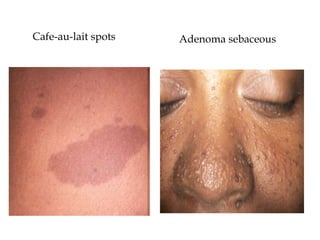
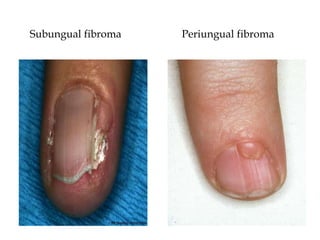
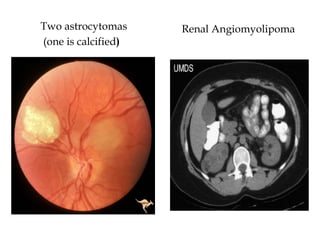
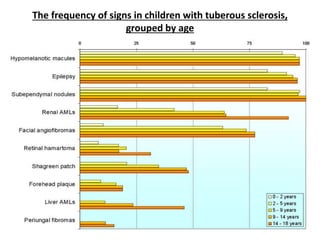
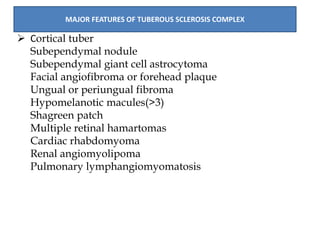


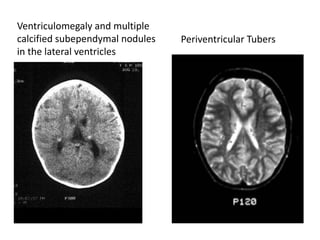

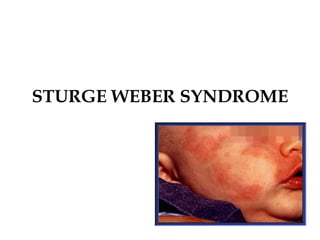


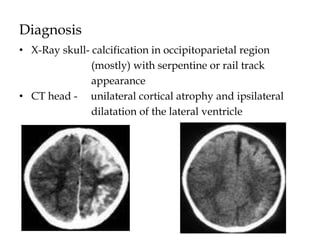




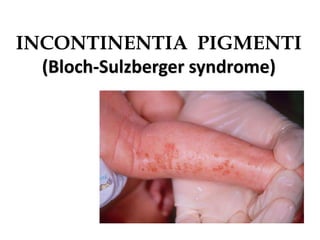

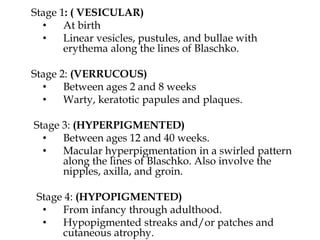








This document provides an overview of several neurocutaneous syndromes: - Neurofibromatosis causes tumors on nerves and affects 1 in 3,000 people. Common features include café-au-lait spots and tumors called neurofibromas. - Tuberous sclerosis causes non-cancerous tumors in many organs and affects 1 in 6,000 people. Common signs include ash-leaf shaped skin lesions and seizures. - Sturge-Weber syndrome is characterized by a birthmark on the face and abnormalities of blood vessels in the brain. It causes seizures and developmental delays.










































































































