
Downloaded 14 times





















The document provides an overview of computational chemistry, focusing on molecular mechanics and dynamics, including their principles, algorithms, and applications. It discusses various computational methods such as semi-empirical techniques, molecular dynamics simulations, and Monte Carlo methods to study molecular structures and interactions. Additionally, it covers key concepts like force fields, energy calculations, and the significance of computer modeling in understanding biochemical phenomena.
Introduction to computational chemistry explaining its use in predicting molecular properties.
Discusses the significance of computational methods in studying complex biological systems.
Covers molecular mechanics basics, including principles, force fields, and energy calculations.
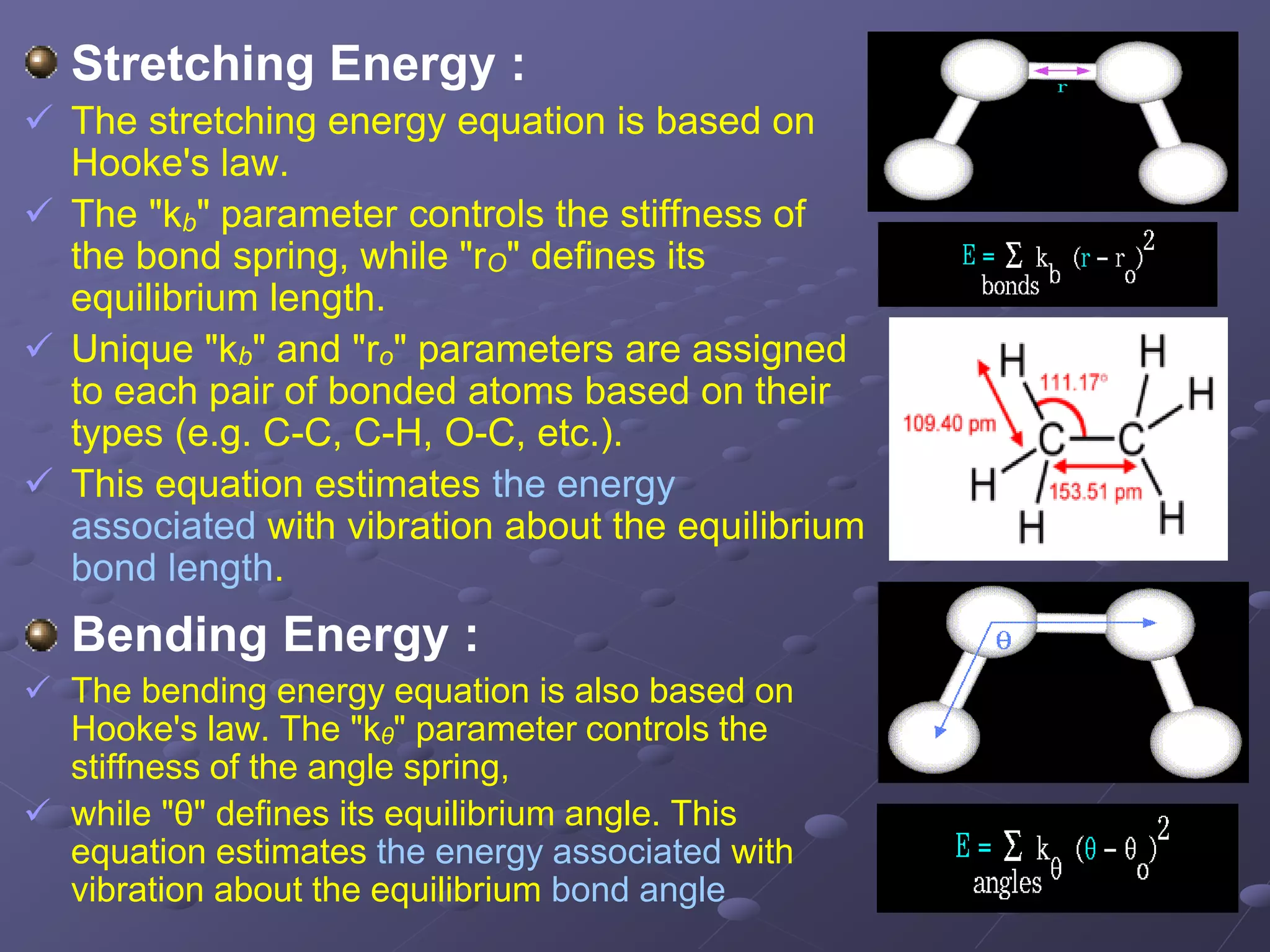
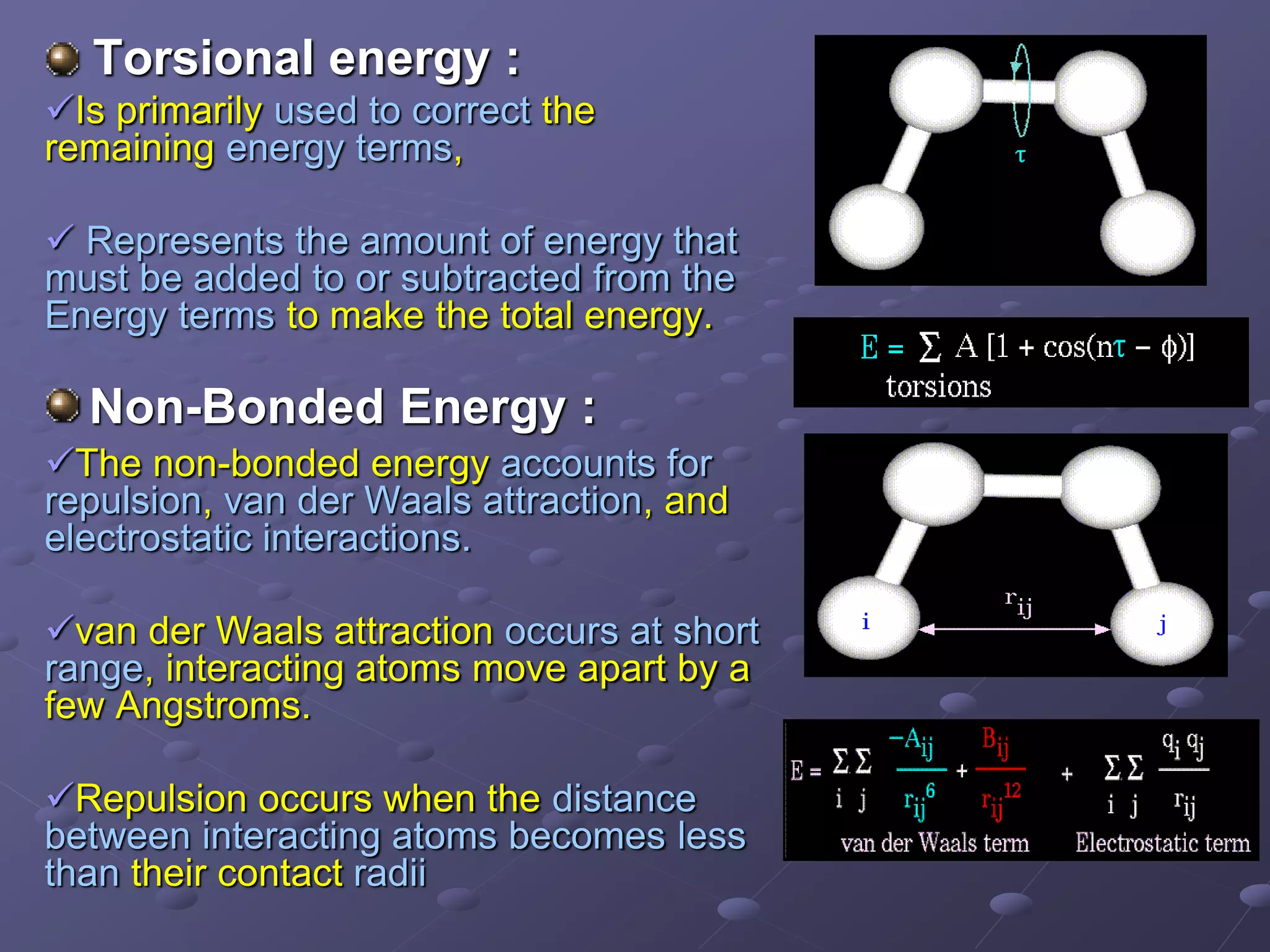
Details the concepts of stretching, bending, torsional, and non-bonded energy in molecular interactions.
Explores various chemical effects like electronegativity and anomeric effects in molecular structures.
Introduces semiempirical methods, their utility, and outlines CNDO, INDO, NDDO, and MNDO methods.
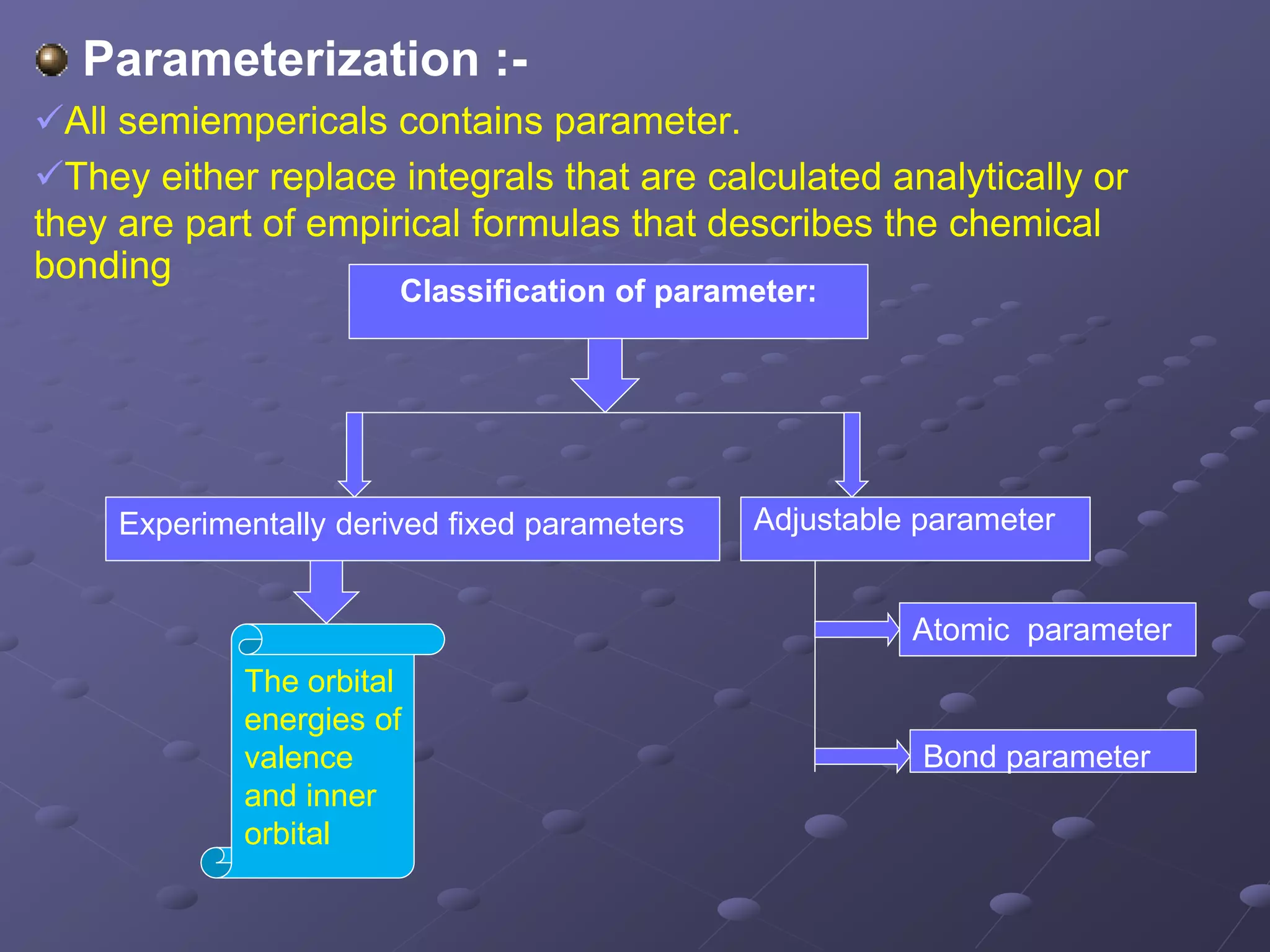
Discusses the role of parameters in semiempirical methods, including fixed and adjustable types.
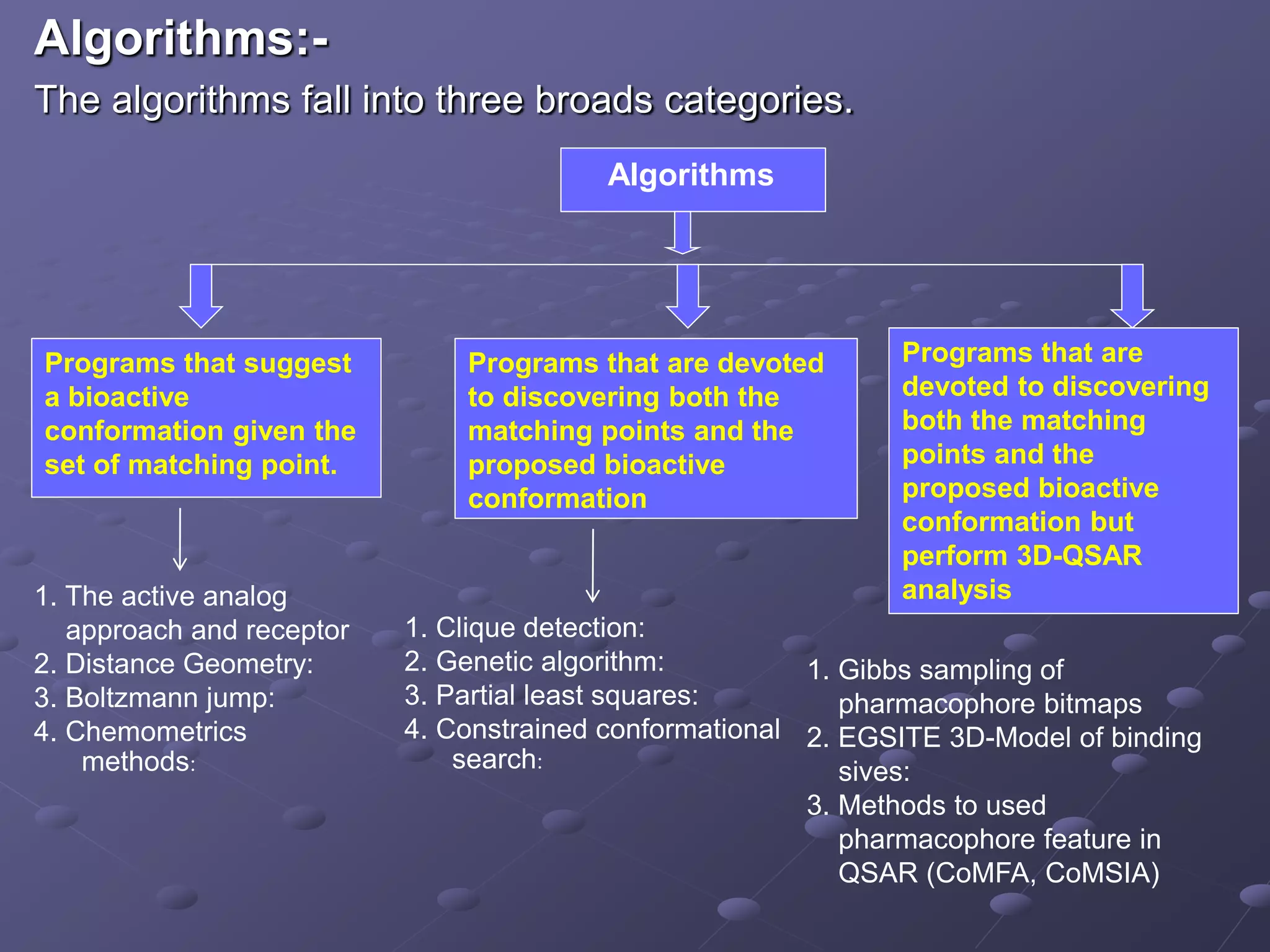
Outlines various algorithms used in computational chemistry for structure identification and optimization.
Introduction to molecular dynamics and its historical context within computational methods.
Explains various MD simulation techniques, their applications, and specific software like AMBER and GROMACS.
Discusses applications of simulations in understanding atomic properties and interpreting biochemical data.
Lists references and sources for further reading on molecular dynamics and computational chemistry.











































































