





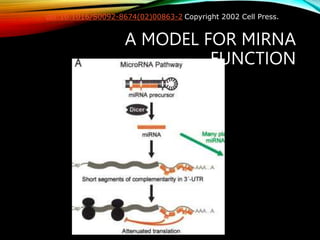


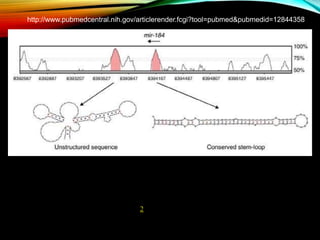


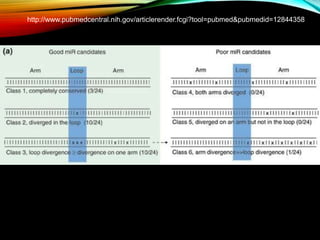








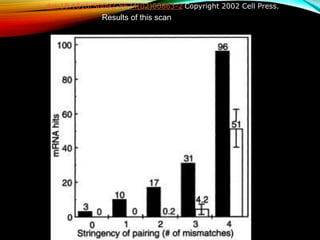





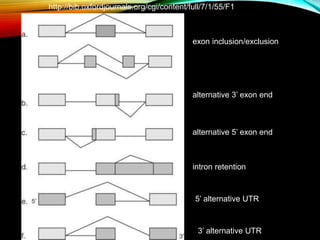


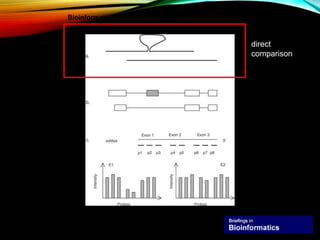




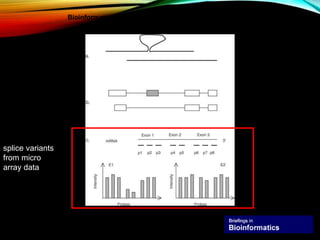


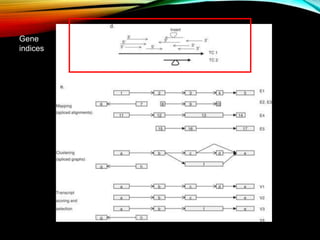


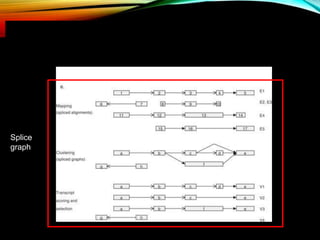



This document discusses two topics: microRNAs and alternative splicing. For microRNAs: Computational methods are used to predict microRNA genes by looking for evolutionarily conserved sequences that can form stem-loop hairpin structures. MicroRNAs regulate gene expression by binding to mRNA. For alternative splicing: Splicing of pre-mRNA can result in different mRNA and protein isoforms through various combinations of exons. Bioinformatics methods aim to identify alternative splice variants by comparing cDNA and genomic sequences and analyzing microarray data. Splice graphs can model alternative splicing pathways.























































































