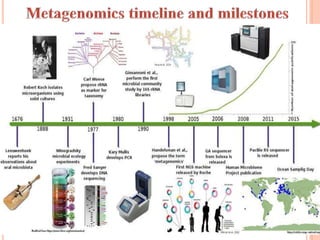
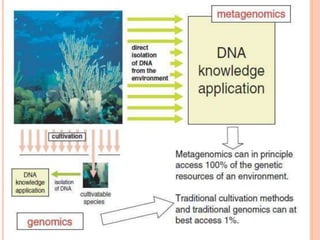
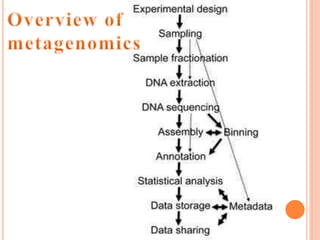
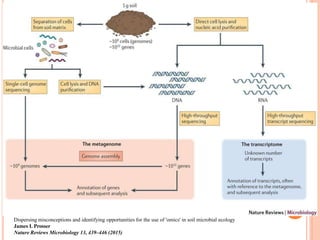
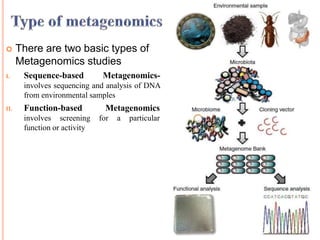
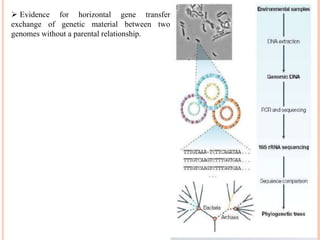
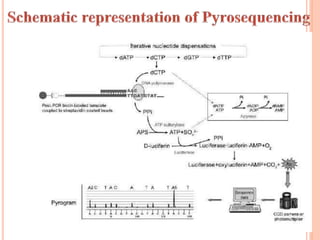
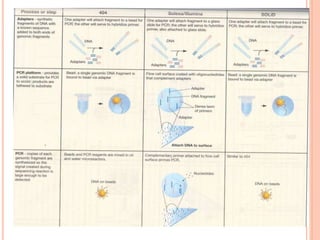
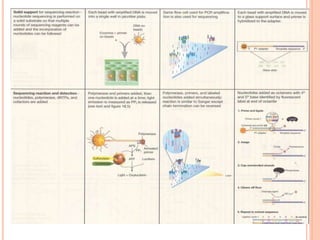
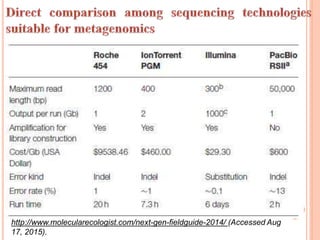
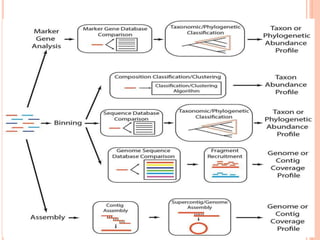
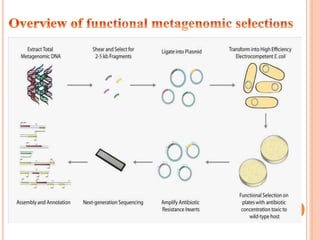
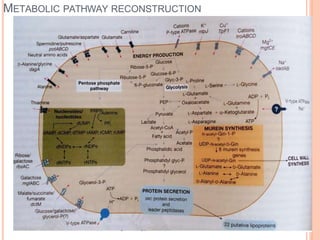
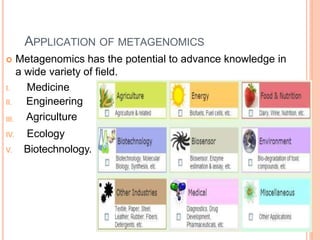
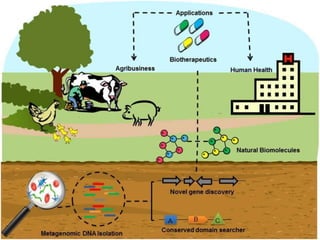
Metagenomics, introduced in 1998, involves the genomic analysis of microorganisms through the direct extraction and cloning of DNA from environmental samples. This field has significant implications in medicine, agriculture, and biotechnology, unlocking uncultured microbial diversity and enabling the discovery of novel genes, enzymes, and pathways. Key methods include sequence-based and function-based metagenomics, which together enhance our understanding of microbial ecology and catalyze advancements in multiple scientific and practical fields.