Downloaded 1,118 times























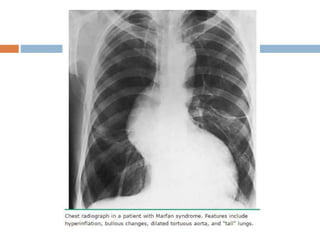

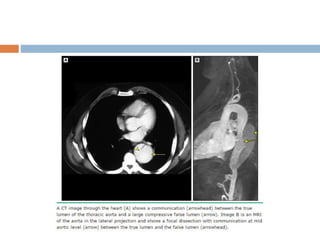






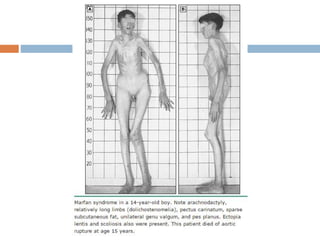



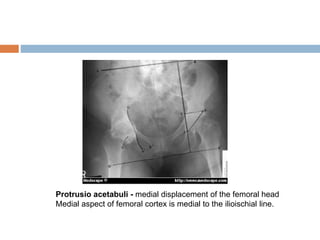


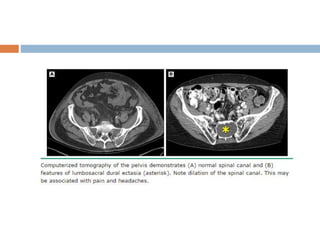







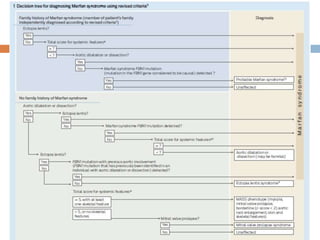
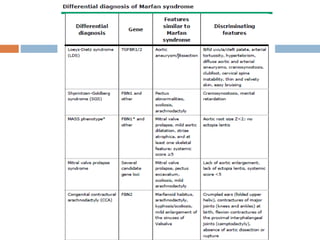
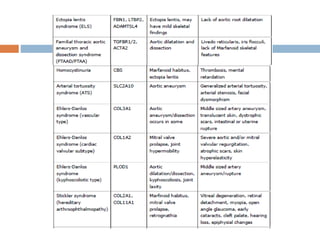
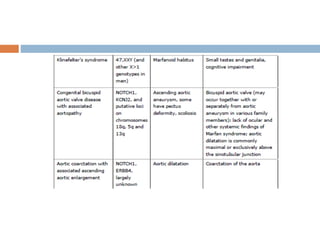






1. Marfan syndrome is an inherited disorder of connective tissue that affects many parts of the body, including the skeletal, ocular, cardiovascular and pulmonary systems. 2. It is caused by mutations in the FBN1 gene which results in abnormal fibrillin protein and connective tissue abnormalities. 3. Diagnosis is based on assessments of the skeletal, ocular, cardiovascular and other body systems compared to established diagnostic criteria such as the Ghent nosology, with a focus on assessments of the aorta and lenses.





























































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)


