Download as PDF, PPTX




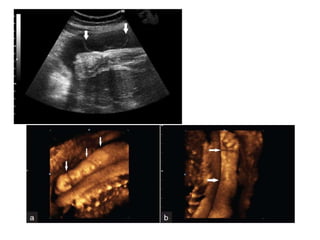

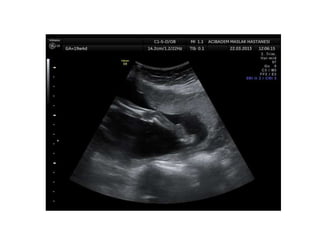
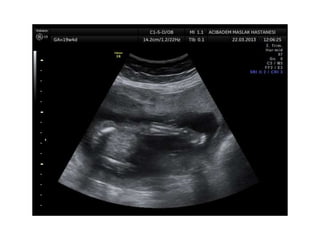

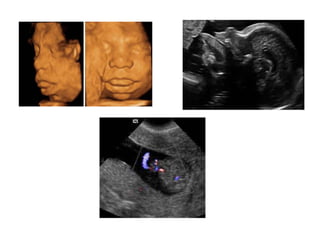


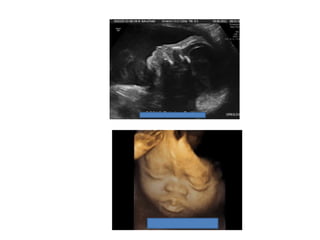

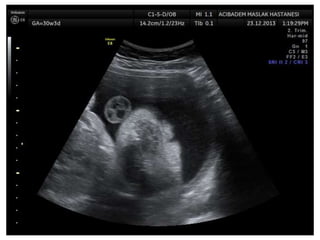
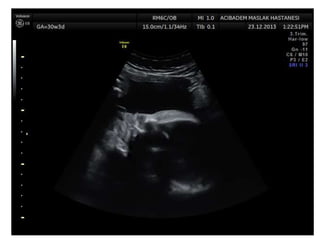
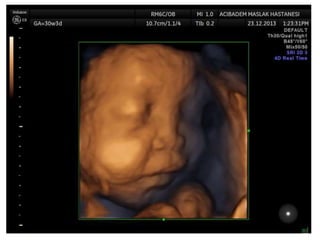
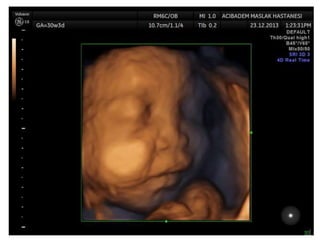

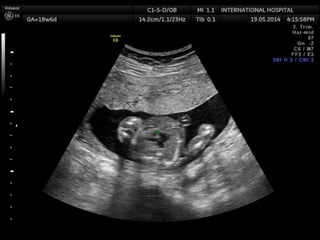
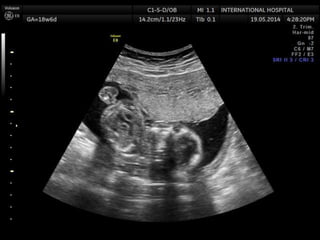
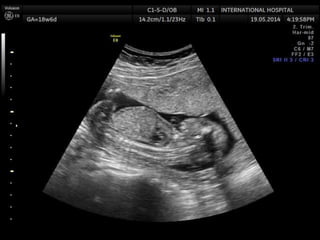
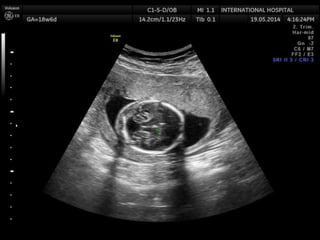
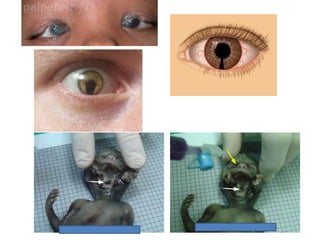

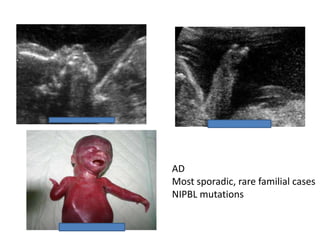

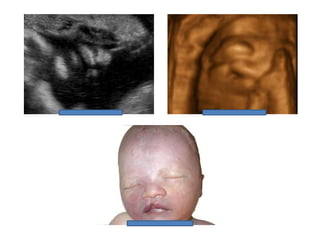
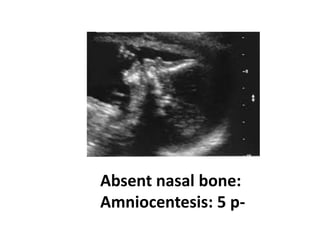

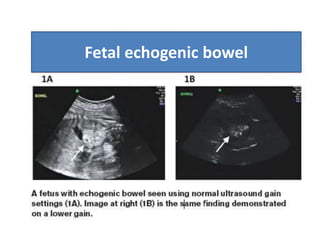
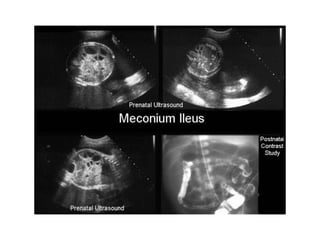
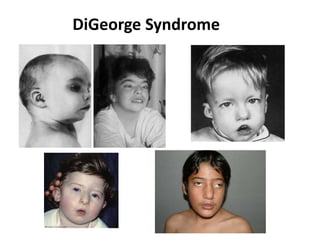

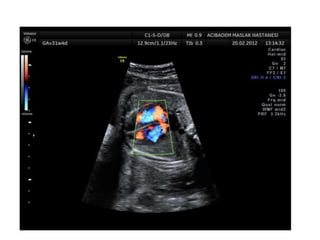
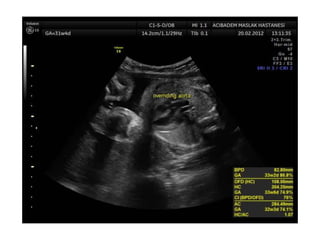
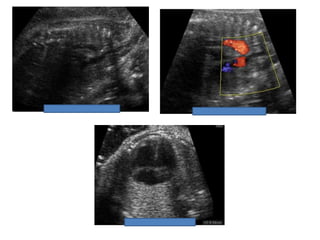

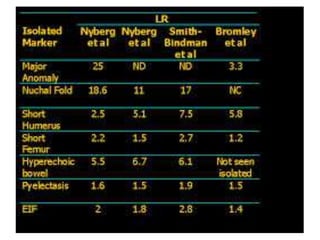
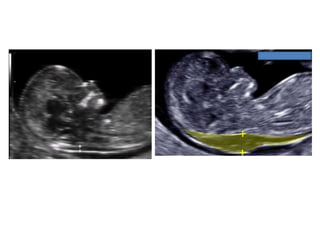

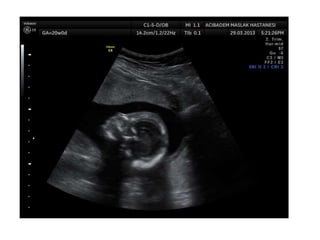
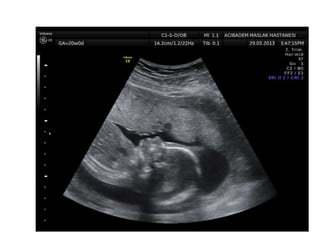
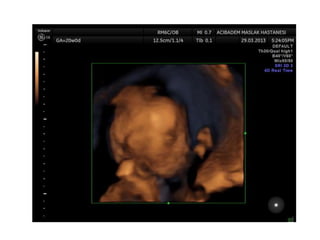
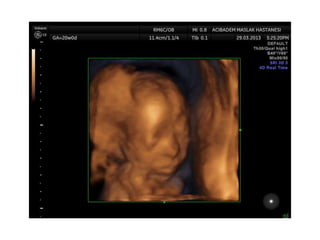
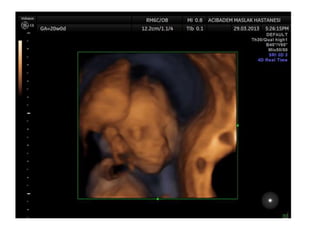
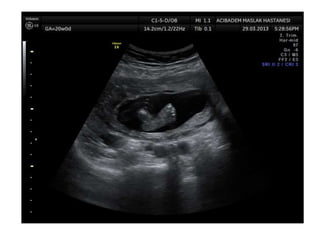
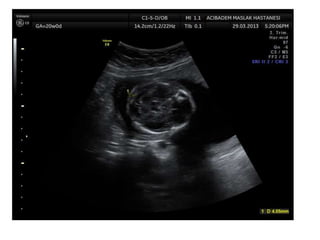


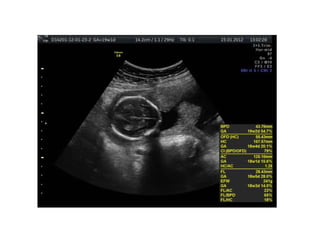
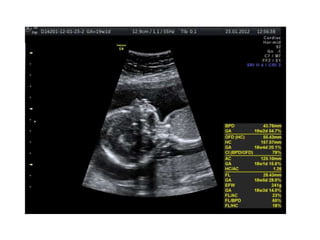
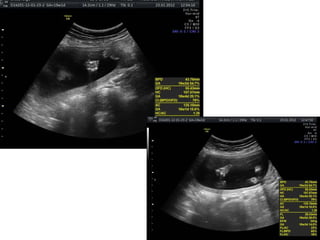
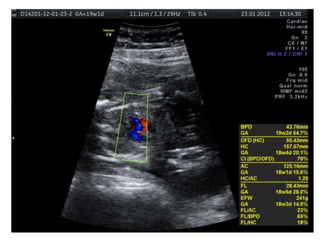
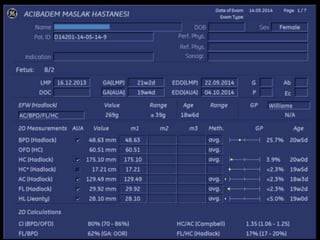
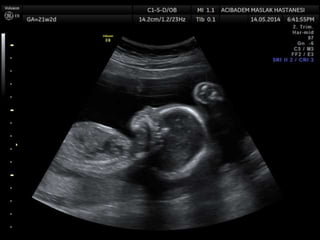
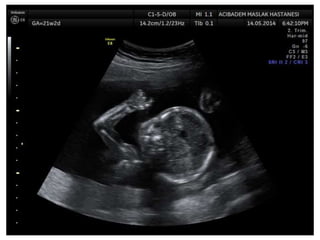
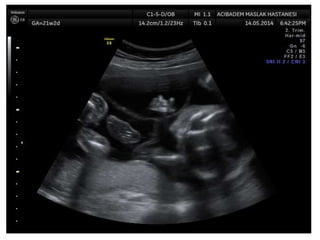
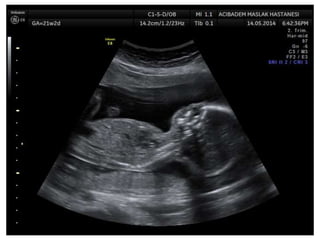

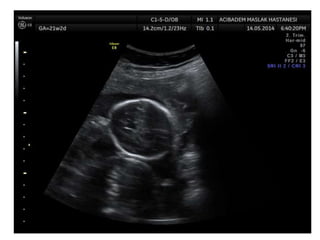

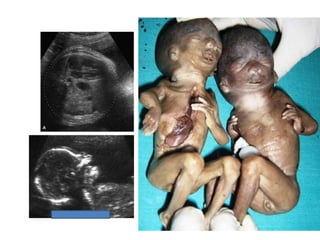

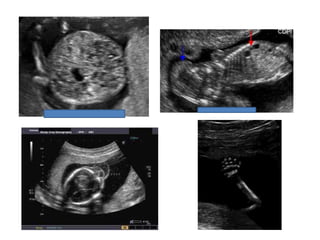


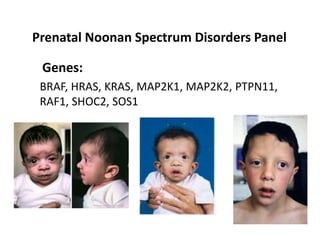
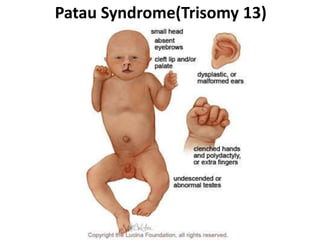



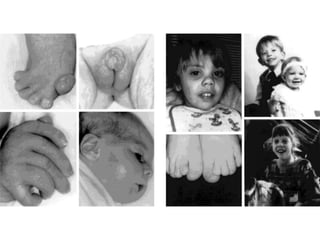


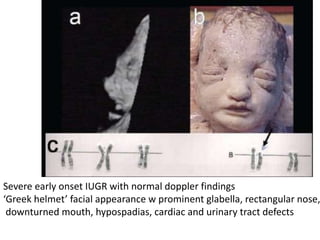
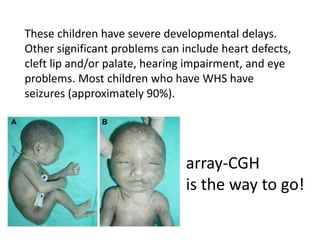


Prenatal diagnosis of frequently seen fetal syndromes can be made based on characteristic ultrasound findings and further confirmed through genetic testing. For Noonan syndrome, mutations in genes such as PTPN11, KRAS, and RAF1 can be found in 17% of cases with increased nuchal translucency and other ultrasound abnormalities. Genetic testing for these and other genes associated with Noonan spectrum disorders can provide prenatal diagnosis. Chromosomal microarray analysis may also detect copy number variants not seen by karyotyping in some cases with abnormal cardiac ultrasound findings and a normal karyotype.























































































