Downloaded 135 times
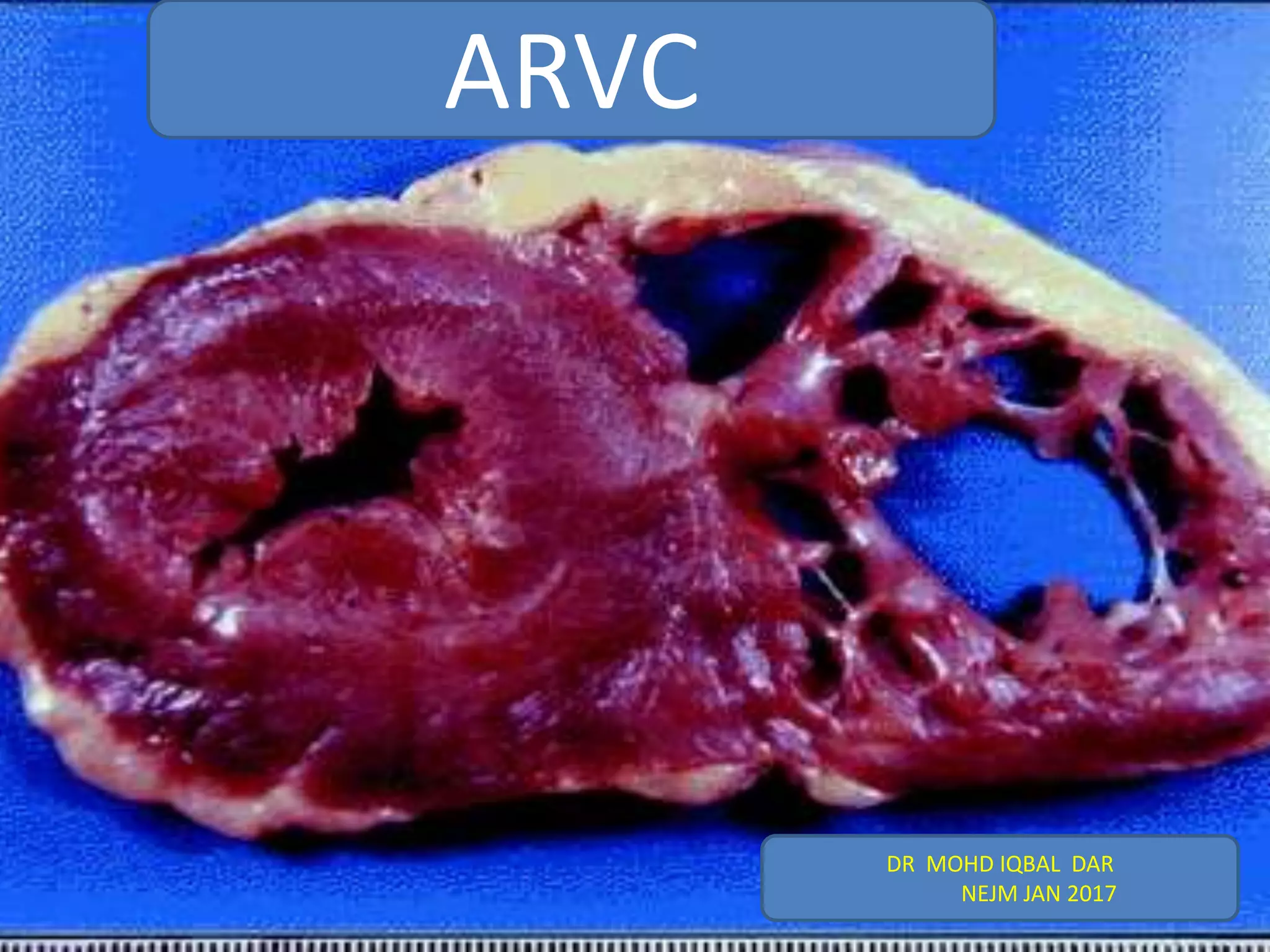


















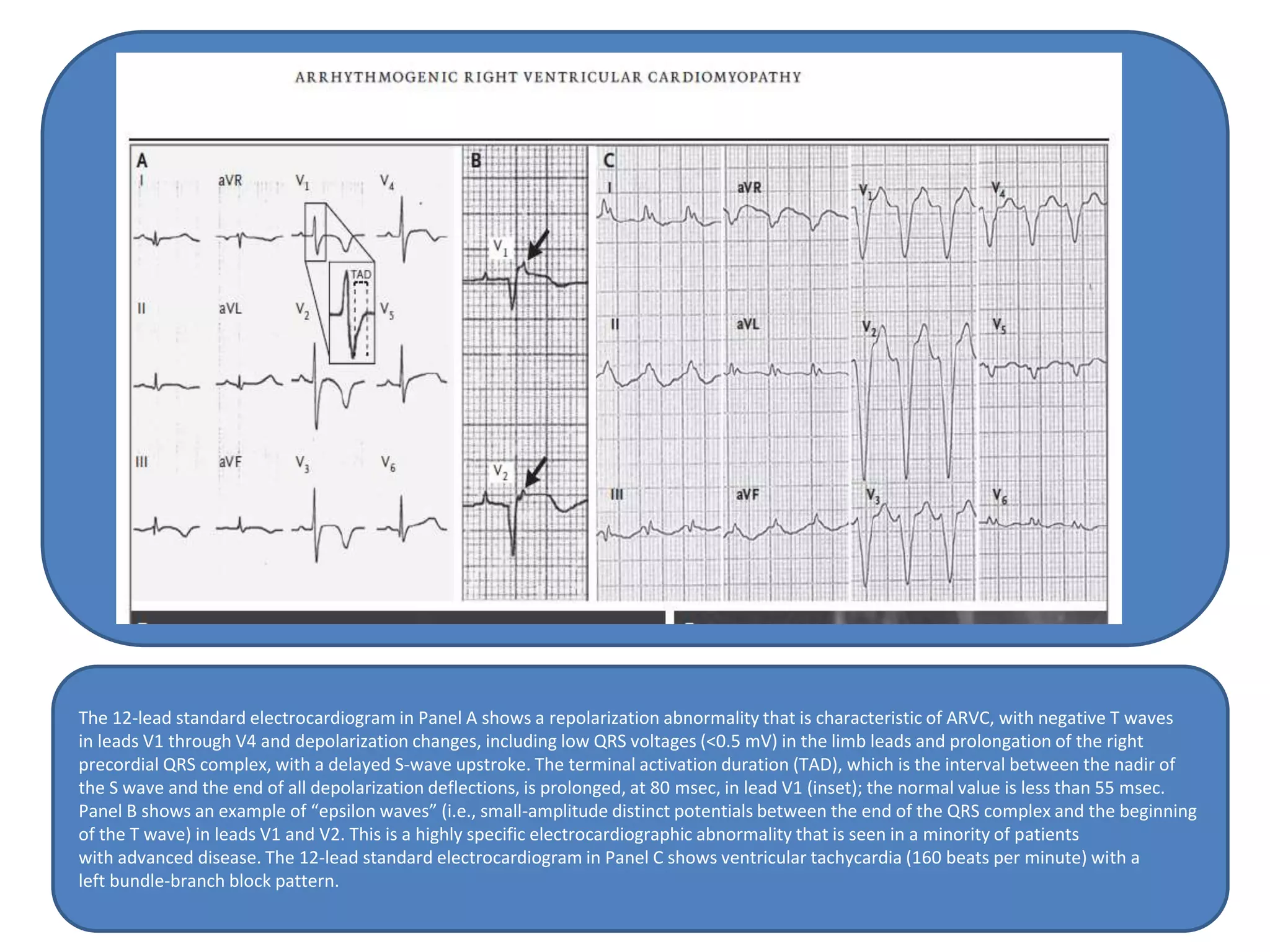

























ARVC is a heritable heart muscle disorder that predominantly affects the right ventricle. It is caused by genetic defects in cardiac desmosomes, which are important for cell-to-cell adhesion. This leads to progressive loss of right ventricular myocardium and replacement by fibrofatty tissue. ARVC can cause dangerous ventricular arrhythmias and is a leading cause of sudden cardiac death in young people. Diagnosis involves imaging tests and electrocardiography to detect right ventricular structural abnormalities and arrhythmias.




































![ARVC and flecainide case report[EI] Jim.docx.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/finalarvcandflecainidecasereporteijim-230918192356-cebc27e5-thumbnail.jpg?width=640&height=640&fit=bounds)



































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)










