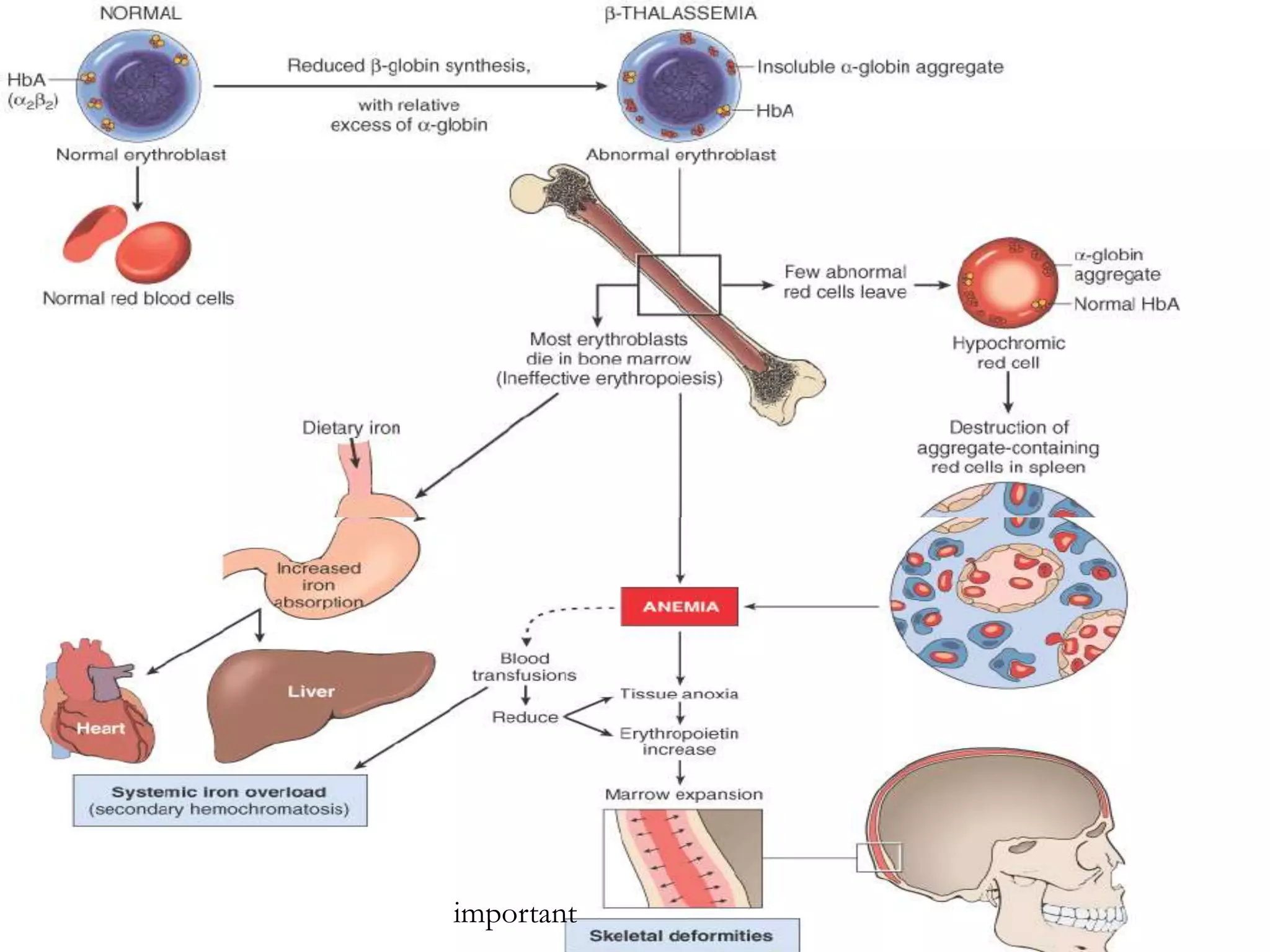
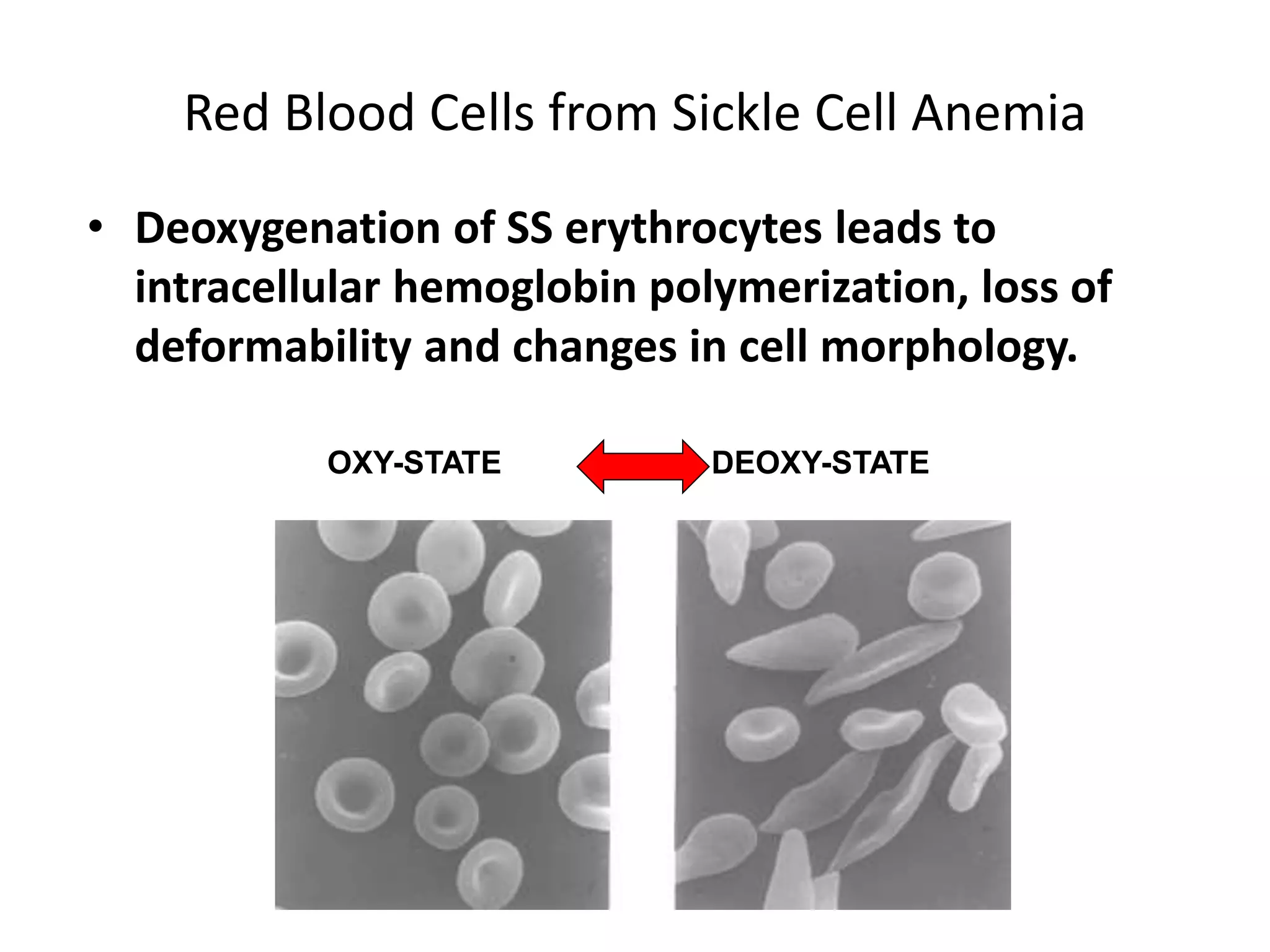
This document discusses thalassemia and sickle cell disease. It begins by describing the different types of hemoglobin present during development and in adults. It then covers the two main types of hemoglobinopathies - qualitative involving abnormal hemoglobin structure like sickle cell, and quantitative involving reduced hemoglobin production like thalassemia. The document goes into detail about the genetic causes and clinical manifestations of alpha and beta thalassemia as well as sickle cell disease. Laboratory findings and treatments are also summarized.