Downloaded 887 times





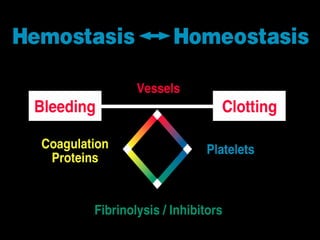
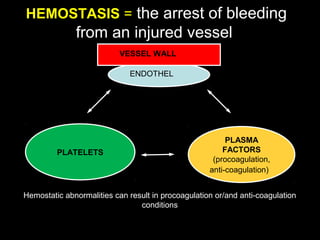
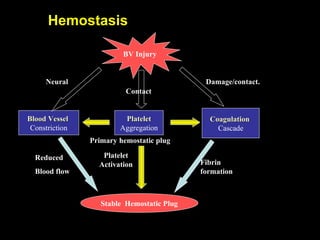

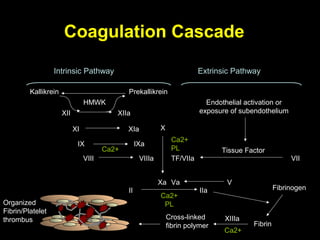
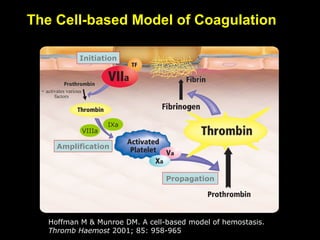

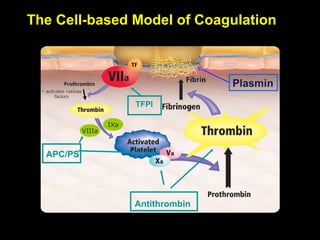

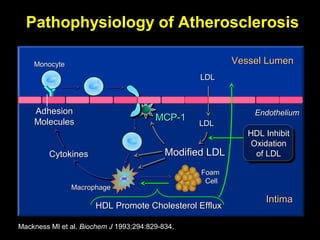
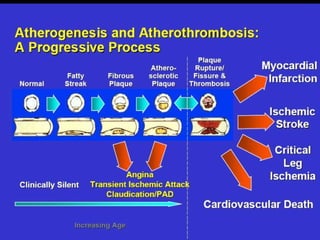



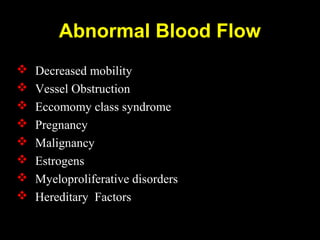



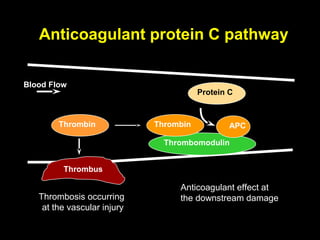
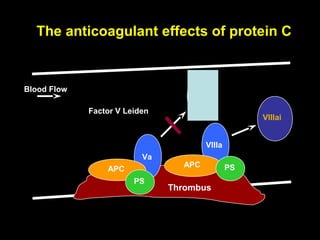







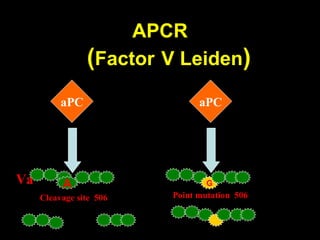






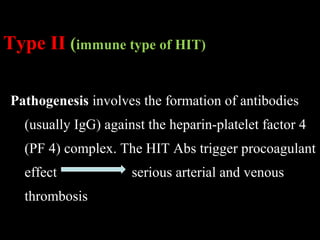
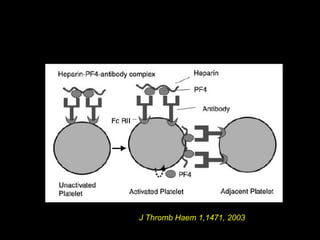


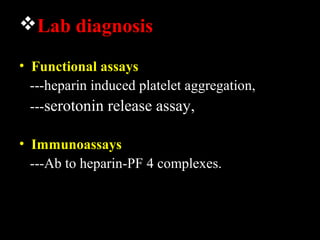

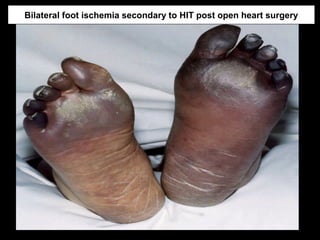
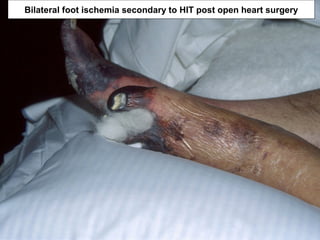
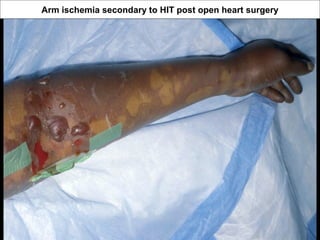











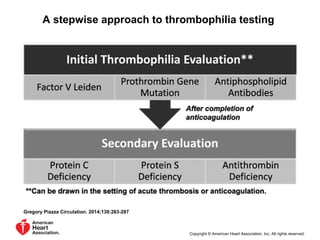







This document discusses hypercoagulable states (thrombophilia). It presents two case studies of patients presenting with deep vein thrombosis (DVT). It then defines thrombophilia as a disorder associated with an increased tendency to form blood clots. The document reviews hemostasis and coagulation mechanisms, inherited and acquired risk factors for hypercoagulability, and recommends a stepwise approach to thrombophilia testing that considers the clinical scenario and implications of testing.



































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)












![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)





