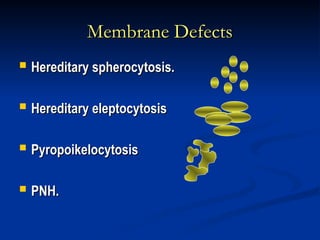
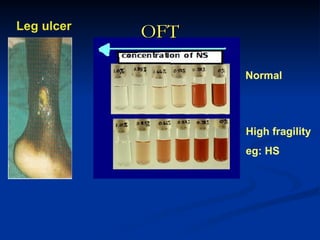
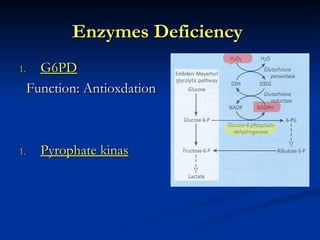
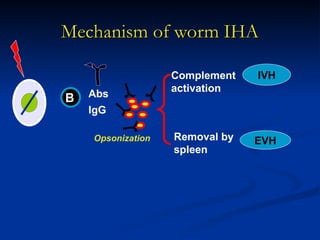
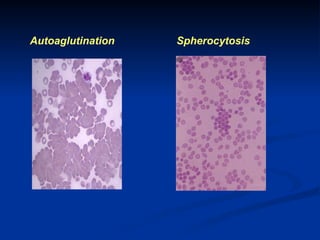
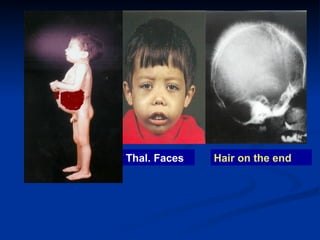
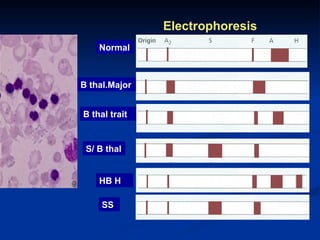
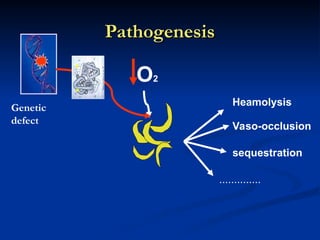
Hemolytic anemias are characterized by premature destruction of red blood cells (RBCs) and can be classified into congenital/acquired and acute/chronic types, with various intrinsic and extrinsic causes. Clinical features may include jaundice, splenomegaly, and black urine, while lab tests reveal abnormalities in RBCs, reticulocyte counts, and cellularity in bone marrow. Treatment options vary based on the underlying condition, including supportive care, immunosuppressive drugs, and potential genetic therapies.