












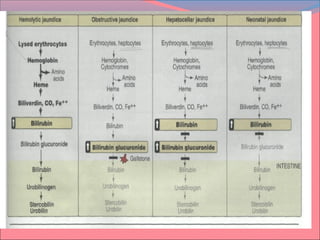
![Most heme from RBCs (85%) - rest from turnover of
cytochromes, p450s, immature erythrocytes.
RBCs last 120 days, degraded by reticuloendothelial
(RE) system [liver and spleen].
Microsomal heme oxygenase hydroxylates methenyl
bridge carbon and oxidizes Fe2+
to Fe3+
. Second
reaction open ring and release methenyl carbon as
CO.
The resulting biliverdin is poorly soluble due to ring
stacking and aggregation.
Serum albumin carries bilirubin in circulation, ligandin
in hepatocytes.](https://image.slidesharecdn.com/must-130522084919-phpapp02/85/HEME-METABOLISM-MUHAMMAD-MUSTANSAR-24-320.jpg)









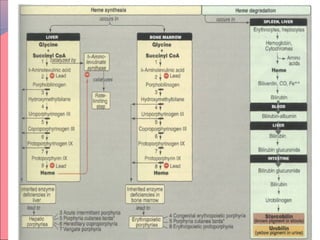
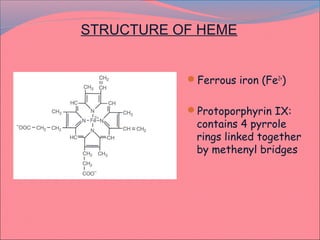
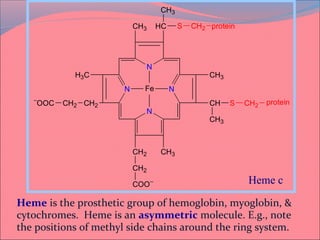
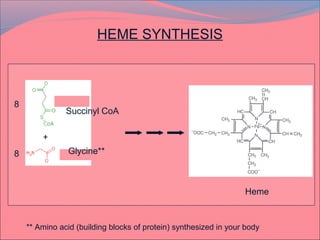
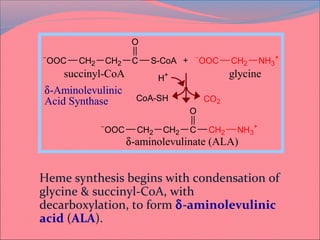
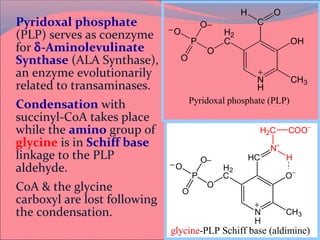
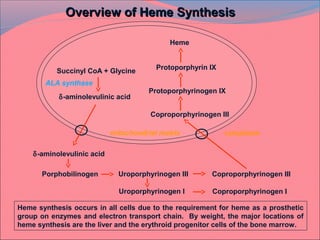
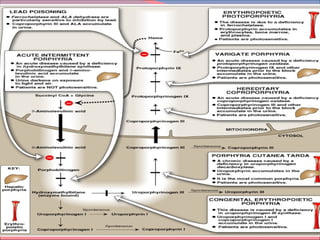
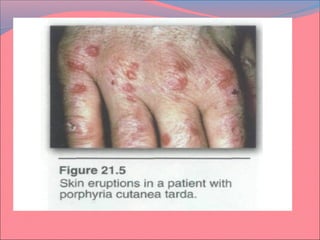
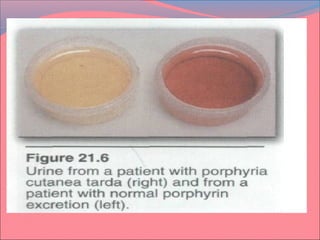
Heme is synthesized from glycine and succinyl-CoA in the liver and red blood cells. The first step is the formation of δ-aminolevulinic acid (ALA) by ALA synthase. ALA then undergoes several modifications to form uroporphyrinogen and coproporphyrinogen. These are converted to protoporphyrinogen IX and then protoporphyrin IX, where iron is inserted by ferrochelatase to form heme. Heme is then used in hemoglobin, myoglobin, cytochromes, and other proteins. Porphyrias are disorders of heme synthesis that cause accumulation of intermediates and are classified as hepatic or erythropoietic











































































































