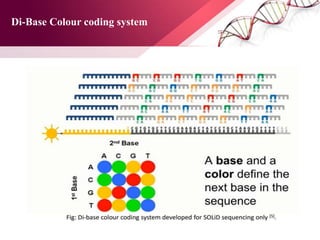
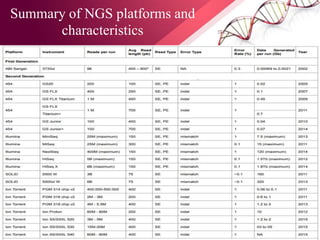
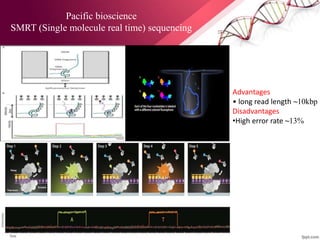
The document presents an overview of next-generation sequencing (NGS) technologies and their applications in crop improvement, highlighting advancements in sequencing methods and their impact on molecular biology. It covers various NGS platforms, their advantages and disadvantages, and detailed applications such as genome sequencing, transcriptome analysis, and epigenetic studies. The document concludes with the future potential and ongoing challenges in utilizing NGS for genetic research and crop enhancement.




















![REGULATORY PROTEIN BINDING DOMAIN
PREDICTION
• Chromatin immunoprecipitation assisted with NGS sequencing is
an efficient method to study genome-wide DNA protein interaction
profile [Varshney et al., 2009].
• The arrival of more efficient NGS techniques have surpassed the
microarray based ChIP-chip, SAGE and STAGE (sequence tag
analysis of genomic enrichment) methods earlier used in such
studies.
• ChIP-seq generates tremendous amount of data, which reveal
insights of gene-regulation and epigenetic modification at a
genome-wide scale, after strong bioinformatic analysis.
• ChIP-seq involves precipitation of DNA with specific antibodies
against the target histone protein or a transcription factor (TF) and
then the precipitated DNA is isolated for further NGS analysis.
• Analysis of the sequence reads provide information about the
target site of specific histone protein or TF on a genome-wide
scale.](https://image.slidesharecdn.com/anjalippt-190911051346/85/Next-generation-sequencing-technologies-for-crop-improvement-47-320.jpg)
![METAGENOMIC ANALYSIS
• Metagenomics is genomic analysis of whole microbial
communities by isolating DNA directly from the environmental
samples.
• This involves preparation of a shotgun or metagenomic library
and sequencing followed by complex data analysis.
• Metagenomics gives new insights to view microbial
populations as many unculturable microbes could not be
studied before using conventional methods [Knief, 2014].
• The various high throughput NGS techniques enable the deep
sequencing of the metagenome, making the identification of
less-abundant microorganisms possible.](https://image.slidesharecdn.com/anjalippt-190911051346/85/Next-generation-sequencing-technologies-for-crop-improvement-48-320.jpg)

![SINGLE CELL GENOMICS
• Improvements in single cell isolation, whole genome amplification
and NGS techniques make it possible to sequence a single cell
genome.
• Although problems associated with the whole genome amplification is
still a challenge, the upcoming TGSTs capable of single molecule
sequencing will make SCGS more accurate and fast [Macaulay and
Voet, 2014].](https://image.slidesharecdn.com/anjalippt-190911051346/85/Next-generation-sequencing-technologies-for-crop-improvement-50-320.jpg)
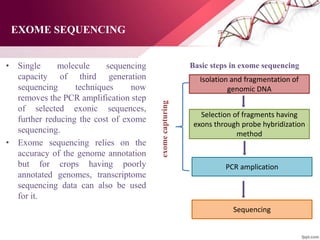
![EXOME SEQUENCING
• Exome referred to the whole set of exons situated in all the
genes (including protein coding and protein non-coding genes)
present in the genome and represent small portion (1–2 %) of it.
• Exome sequencing can provide information of variants present
in coding region of the genome of a large number of
individuals with deep coverage and more cost-effective manner
[Warr et al., 2015].
• Exome sequencing is a two-step process.
1. Exome capturing
2. Sequencing](https://image.slidesharecdn.com/anjalippt-190911051346/85/Next-generation-sequencing-technologies-for-crop-improvement-51-320.jpg)