Downloaded 73 times











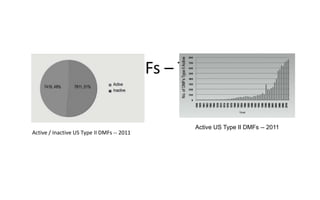







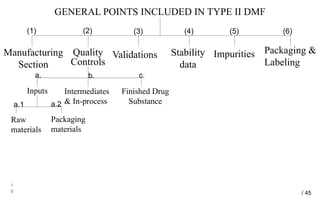










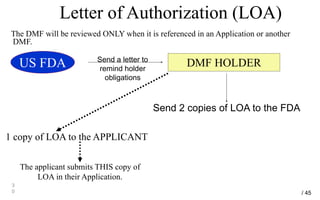




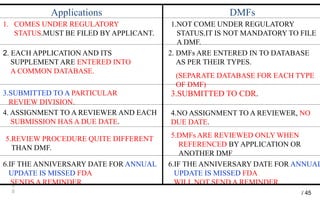












Drug Master Files (DMFs) provide confidential information to regulatory agencies to support drug approvals. There are different types of DMFs for drug substances, products, packaging, and excipients. DMFs are submitted voluntarily to the FDA and are kept confidential. They are reviewed only when referenced in an application such as an NDA. Applicants submit a Letter of Authorization to the FDA and DMF holder to access the DMF information during review. DMF holders must provide annual updates to keep the information current.





















































![Drug master file ppt [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/drugmasterfilepptautosaved-200130192621-thumbnail.jpg?width=640&height=640&fit=bounds)





















































