Download as PDF, PPTX













![14
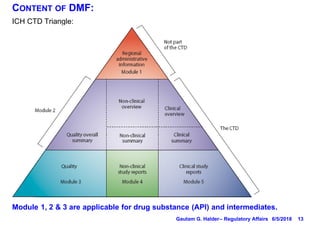
MODULE 1 CONTENTS
Module 1. Administrative Information and Prescribing Information
Module 1 should contain the following information
ü Cover Letter
ü Administrative Information
§ Addresses of DMF Holder and Manufacturing & Testing facilities
§ Responsible and Contact persons
ü Statement of Commitment
ü Generic Drug User Fee Cover Sheet (3794), where applicable
ü Letter of Authorization, where applicable
ü US Agent Appointment Letter
ü Debarment Certification [Section 306(k)(1)]
ü Environmental Statement
ü Specimen Product Label
6/5/2018Gautam G. Halder– Regulatory Affairs](https://image.slidesharecdn.com/result-180605155259/85/USDMF-Preparation-and-Submissions-14-320.jpg)









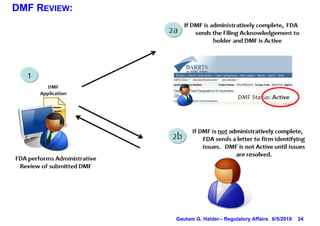


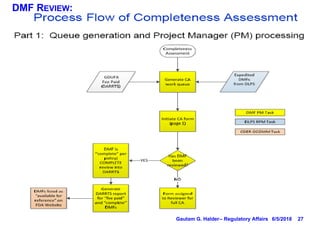
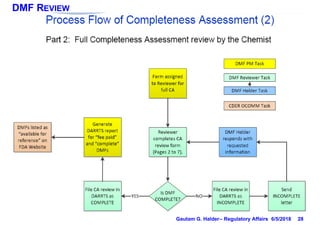






This document provides information on drug master files (DMFs), including: - There are 5 types of DMFs which contain information on drug substances, packaging materials, excipients, and other reference materials. - A Type II DMF contains information about drug substances and products, and must be submitted in ICH CTD format. - Modules 1-3 of the CTD are included in a DMF, with Module 1 containing administrative information, Module 2 providing a quality overall summary, and Module 3 containing detailed information on manufacturing, characterization, and controls. - DMF holders must notify authorized parties of any amendments and provide annual reports on the anniversary of the original submission.


































![Drug master file ppt [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/drugmasterfilepptautosaved-200130192621-thumbnail.jpg?width=640&height=640&fit=bounds)
































