Downloaded 308 times




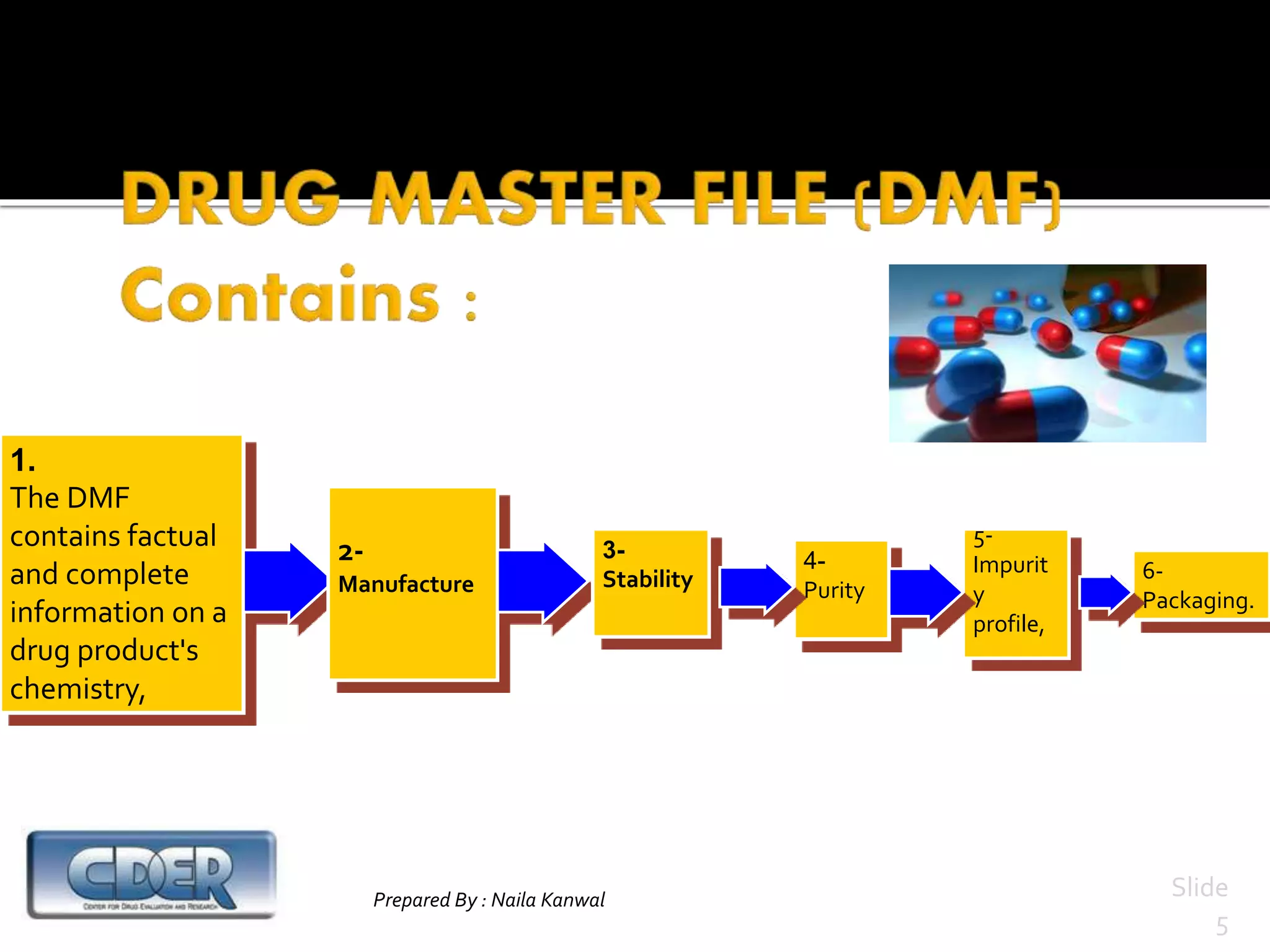





















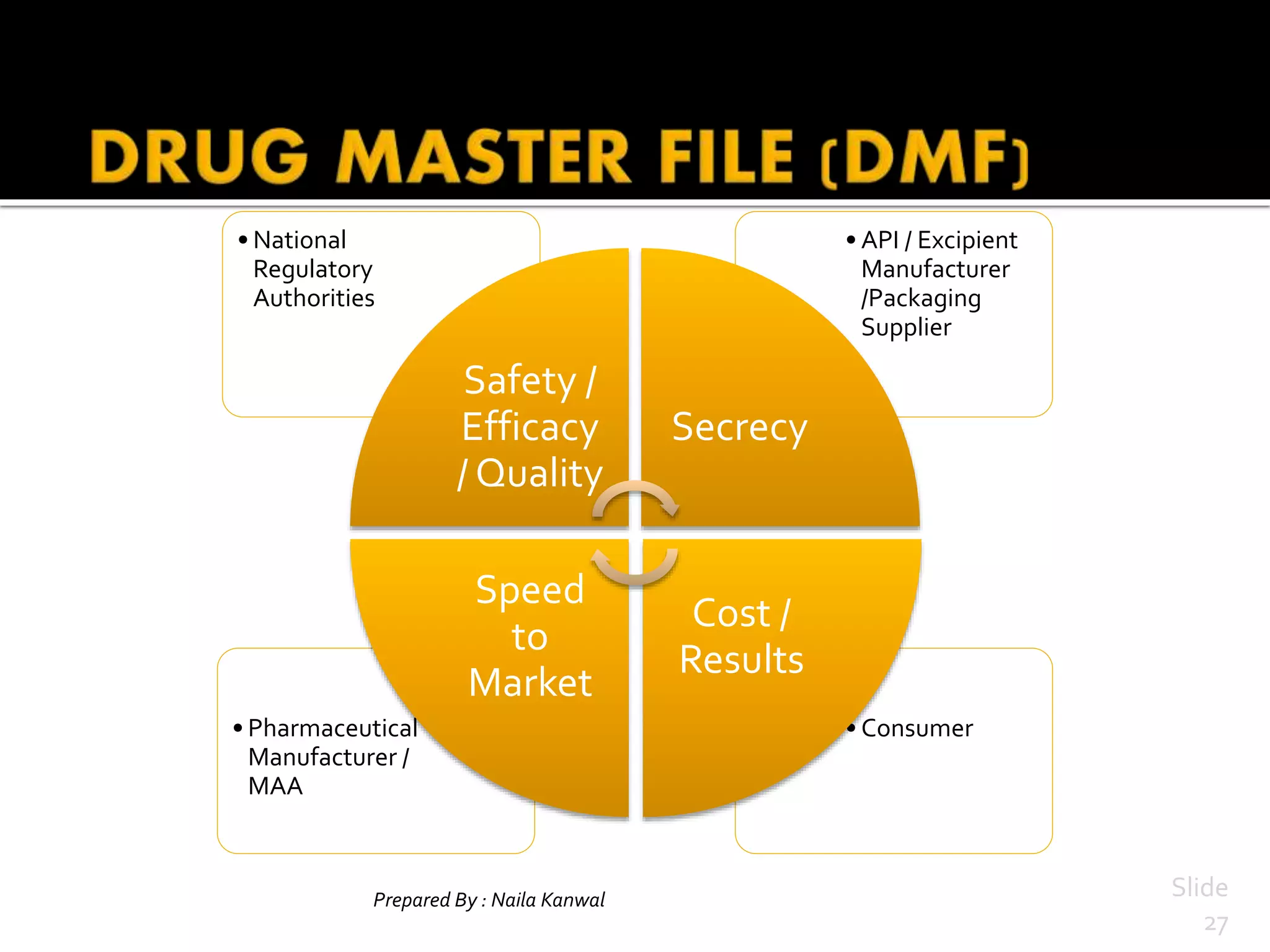


The document discusses drug master files (DMFs), which provide confidential manufacturing and facilities information to regulatory authorities. It covers: 1. DMFs are submitted voluntarily to regulatory agencies and contain detailed information about manufacturing processes, facilities, and components used in drugs. 2. They allow companies to protect intellectual property while ensuring regulatory disclosure, as when two firms partner in drug development or manufacturing. 3. Requirements for DMFs vary globally, with sections in the US, Europe, Canada, and Australia addressed in the presentation. Harmonization efforts aim to standardize DMF processes internationally.

























![European_Union.ppt.Nikhil[1]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803111014-996baf4e-thumbnail.jpg?width=640&height=640&fit=bounds)






















































