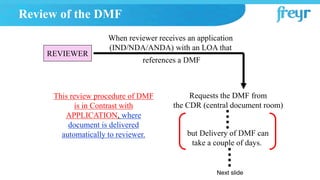
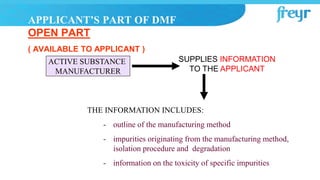
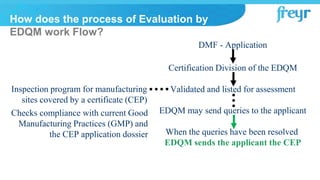
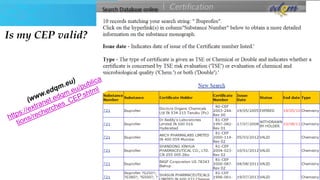
The document provides an overview of Drug Master Files (DMFs) and Certificates of Suitability (CEPs) for drug substances. It describes that a DMF is a confidential submission to regulatory agencies containing manufacturing and quality control information about a substance or component. It discusses the different types of DMFs, how they are submitted and reviewed, annual reporting requirements, and retirement. It also describes that a CEP is issued by EDQM and demonstrates that a substance's purity is controlled according to European Pharmacopoeia monographs, easing approval in Europe.










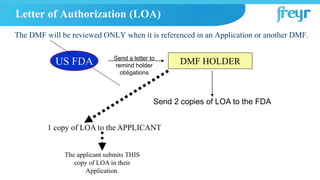
![Letter of Authorization (LOA)
DMF HOLDER
1 copy of LOA to the APPLICANT
The applicant submits THIS
copy of LOA in their
Application.
LOA to the FDA
Key points - LOA
Sending LOA is the only mechanism
which triggers the review procedure of
DMF.
If the holder cross references its own DMF,
the holder should supply following
information in the LOA.
• DMF number
• Specific product(s) covered by the DMF
• Section numbers and/or page numbers to
be referenced
[Authorization to refer to a DMF]](https://image.slidesharecdn.com/40c9bdf0-477b-4a4e-ba9d-257a733c1078-150814132222-lva1-app6892/85/DMF-CEP-11-320.jpg)