Downloaded 106 times










































1) The document discusses submitting electronic DMF/ASMF dossiers to regulatory agencies in the US, Canada, EU, and Switzerland. It defines DMF/ASMF and describes the different types used across regions. 2) Key differences in DMF types and structures between regions are explained, including the division of dossiers into open/applicants parts and closed/restricted parts. CTD sections to be included in each part are provided for examples. 3) Conversion timelines and requirements for electronic submission formats like eCTD are outlined for each region. Canada and EU now require eCTD format for new submissions, while FDA accepts both eCTD and paper.
Introduction and outline of a webinar discussing DMF/ASMF submissions in various regions.
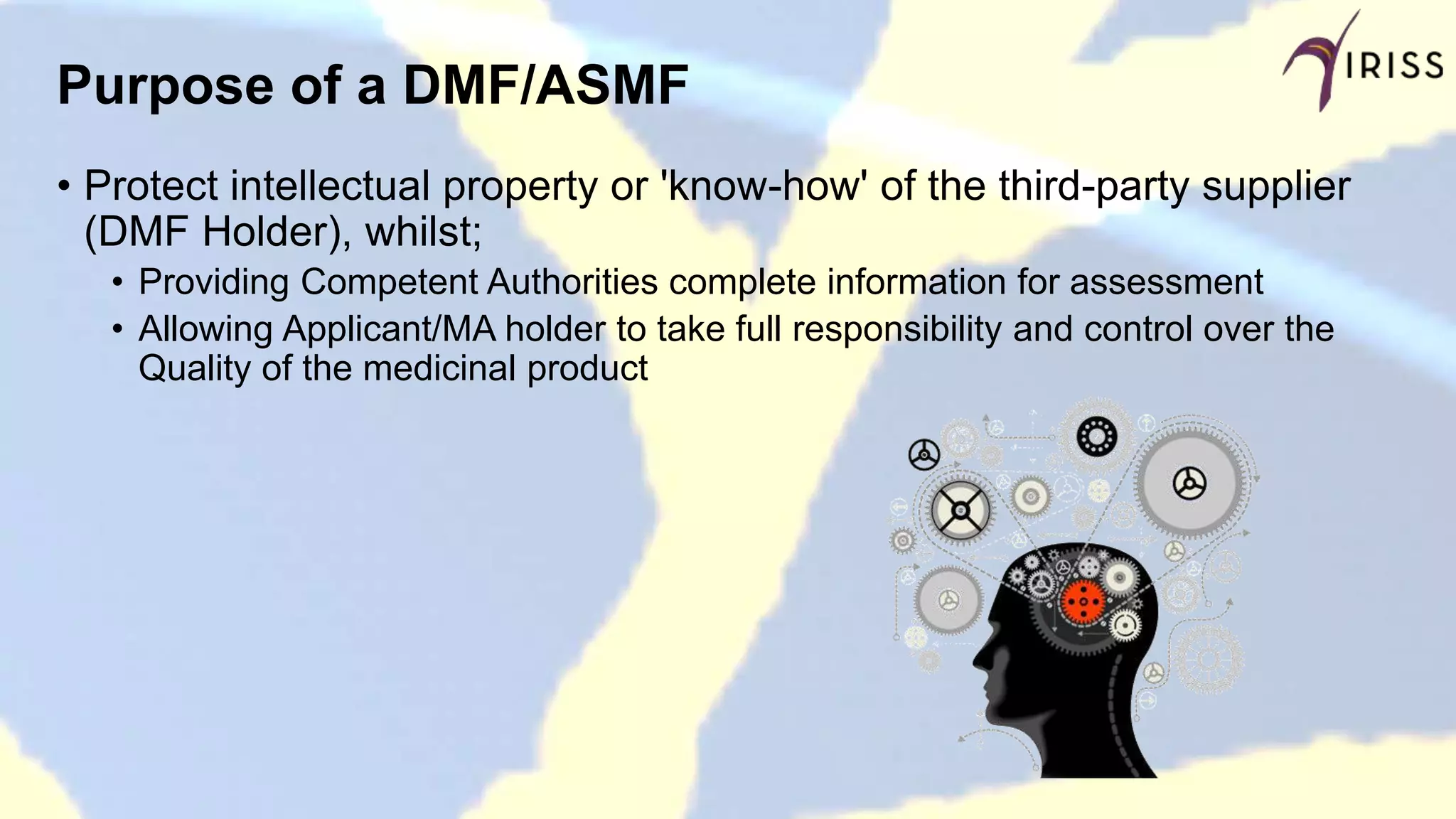
Defines DMF as a CMC submission to ensure confidentiality and support for quality assessment.
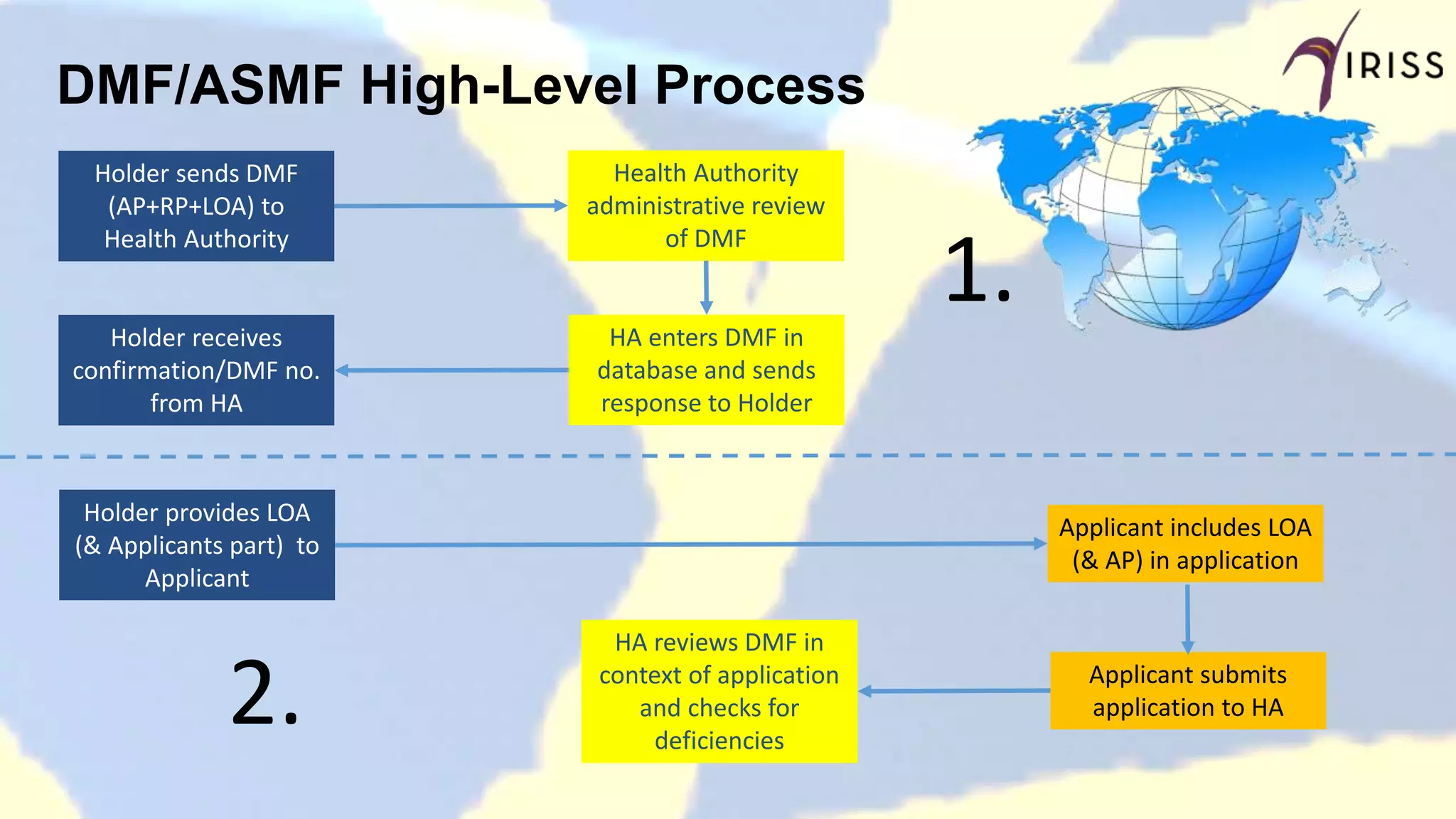
Describes the process flow from DMF submission to health authority review and application integration.
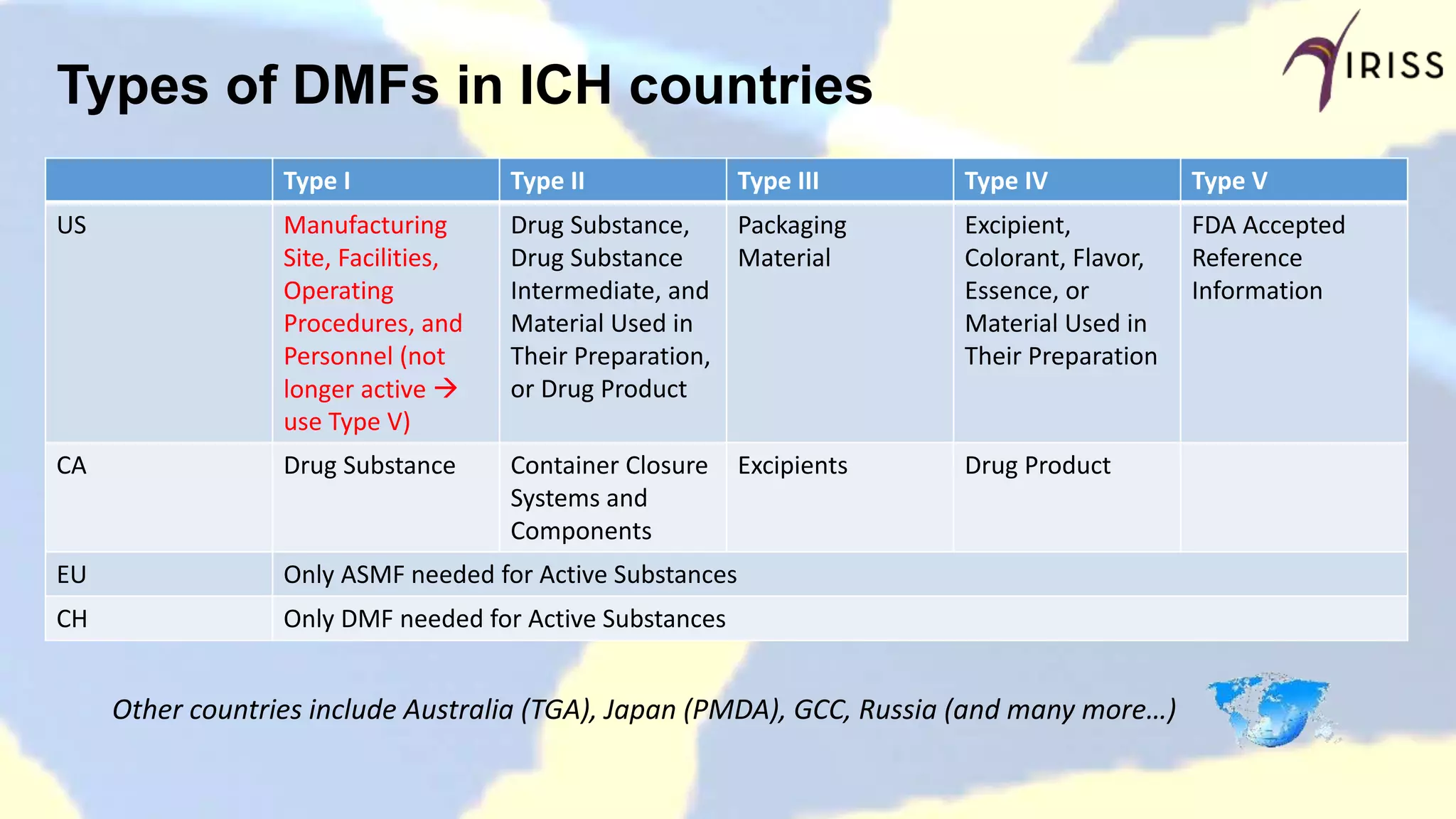
Overview of DMF types across ICH countries, specifically US, CA, EU, and CH.
Details various US DMF types, their characteristics, submission requirements, and FDA review processes.
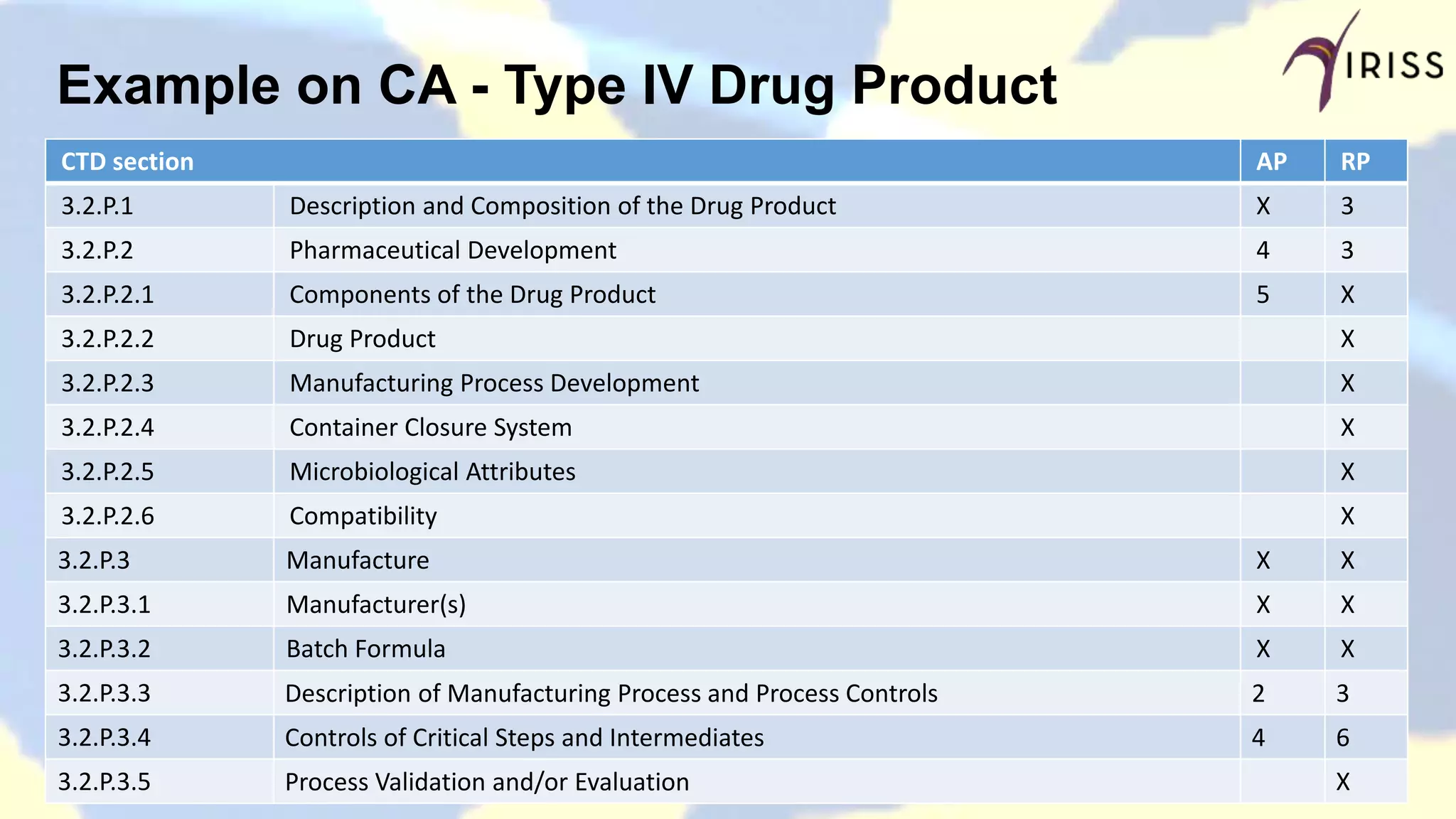
Outlines DMF types, submission guidelines, outcomes, and expectations in Canada.
Discusses the European Active Substance Master File definitions, requirements, and maintenance.
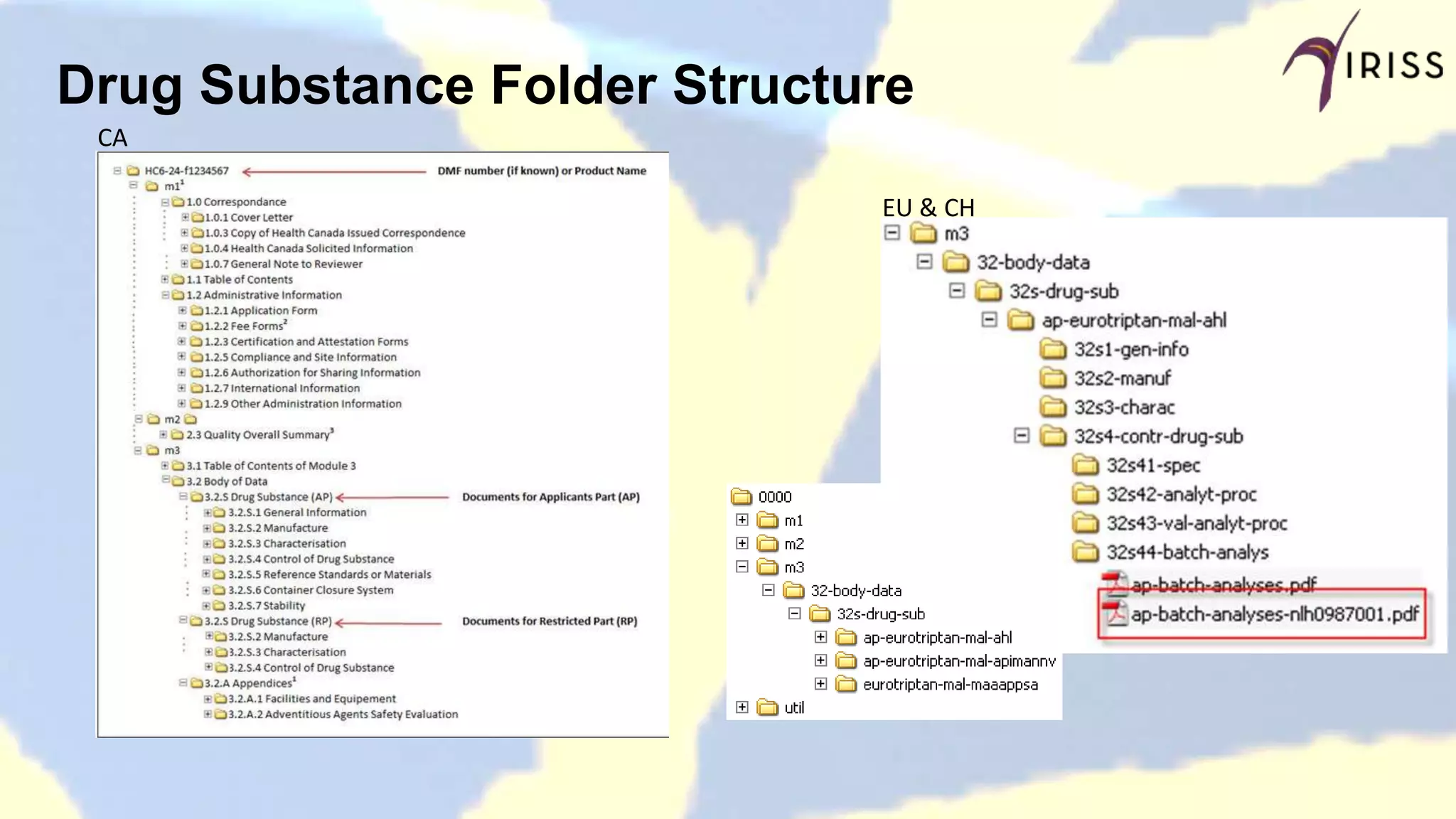
Compares DMF dossier structures and differences between different countries.
Specifies the requirements for summaries included in DMF submissions.
Highlights the formats, methods, and specifications for electronic DMF submissions in various regions.
Summarizes the varying scopes, definitions, review procedures, and emphasizes eCTD adoption across regions.







































































![Rheumatic Fever CASE PRESENTATION [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationautosaved-251123182512-9d9b0da4-thumbnail.jpg?width=640&height=640&fit=bounds)

