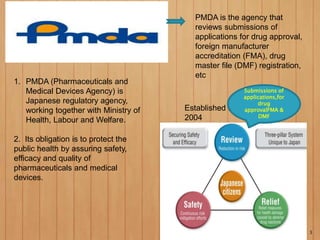
1. The PMDA (Pharmaceuticals and Medical Devices Agency) is the Japanese regulatory agency that reviews submissions for drug and medical device approval to ensure safety, efficacy, and quality. It was established in 2004.
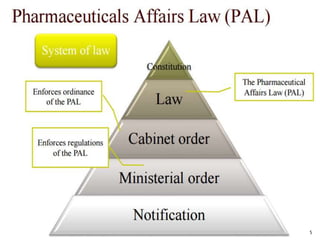
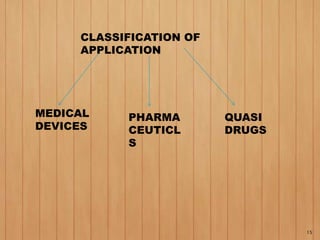
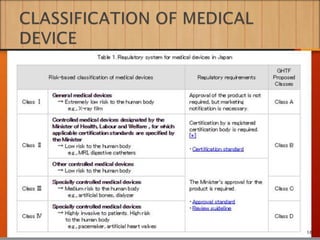
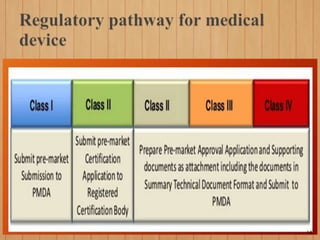
2. To market a drug in Japan, approval must be obtained for each product by demonstrating efficacy and safety through examinations. Foreign manufacturers must be accredited through the FMA process. Medical devices are classified and require pre-market notification, certification, or approval depending on the risk class.
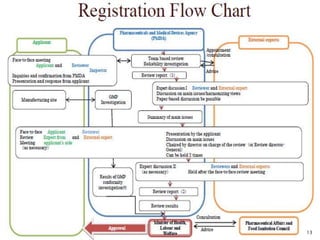
3. Registering products in Japan requires navigating a complex process that can involve clinical trials and high fees. Pursuing product registration requires carefully considering the market demand to determine if pursuing approval is worthwhile.













![An “Applicant” is required to submit “Application for
Accreditation” (Form No. 18 in the PAL Enforcement
Regulations) that is addressed to the Minister, and
“Application for Accreditation Examination” [Form No.16-
(2)]
Documents to Be Attached to Accreditation Application:-
A medical certificate from a physician which indicates
whether or not an “Applicant” has mental disorders or is
addicted to narcotics, cannabis, opium or stimulant drugs.
A curriculum vitae of the person who is responsible to the
manufacturing establishment
21](https://image.slidesharecdn.com/drugapprovalprocessinjapan-170130101148/85/Drug-approval-process-in-japan-21-320.jpg)























![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)






























































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)
