Download to read offline























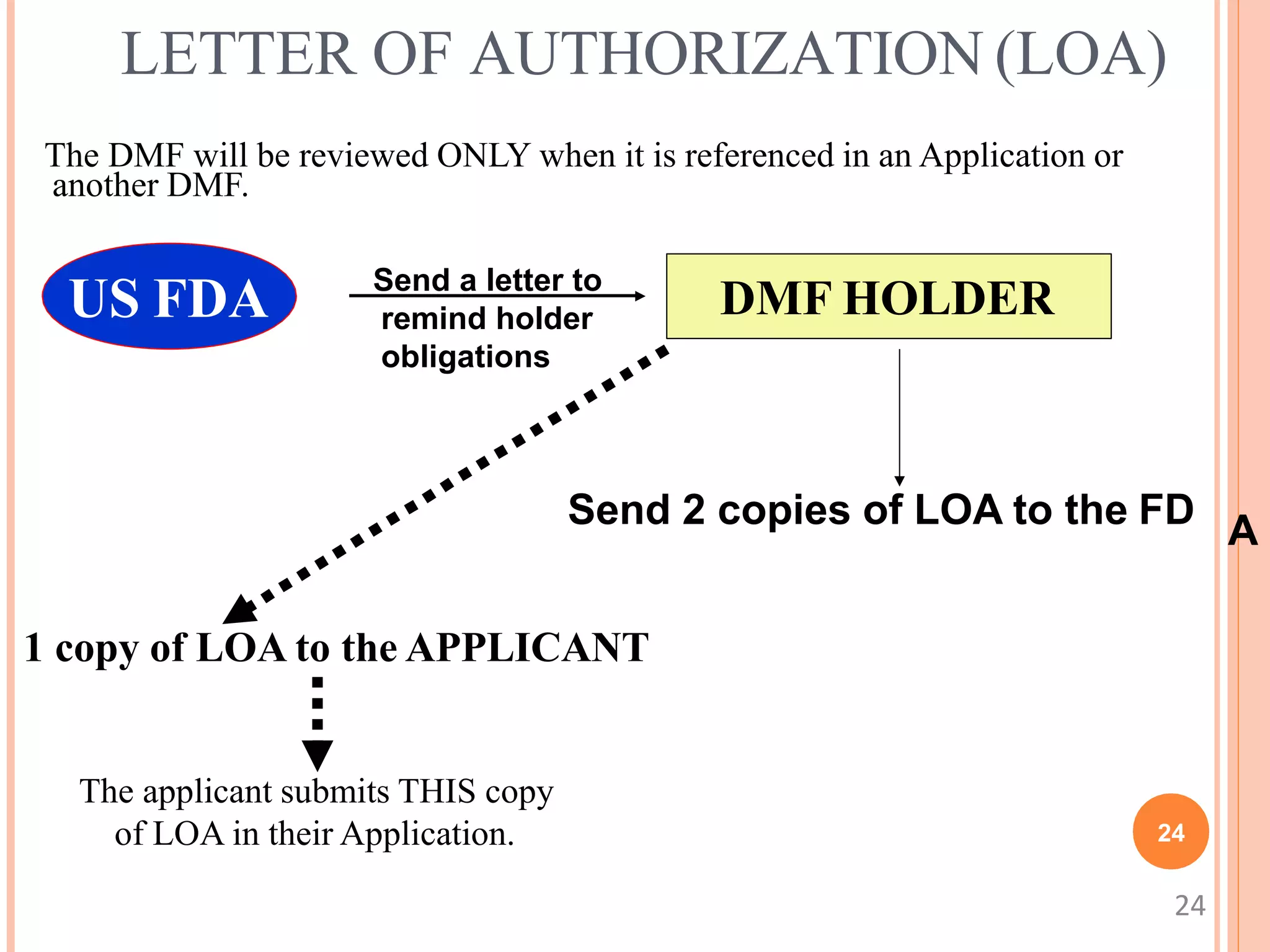



















The document provides information about Drug Master Files (DMFs), including: 1. A DMF contains confidential information about the manufacturing and controls of a drug product or component submitted to the FDA. There are currently four types of DMFs. 2. A DMF is not required to be filed but can provide information to support applications like an IND, NDA, or ANDA. The holder submits the DMF and is responsible for updating it annually or when changes occur. 3. A DMF is only reviewed when referenced by an application through a Letter of Authorization from the holder. The FDA communicates deficiencies only to the holder, keeping the contents confidential.










































![Drug master file ppt [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/drugmasterfilepptautosaved-200130192621-thumbnail.jpg?width=640&height=640&fit=bounds)








































