Downloaded 380 times



























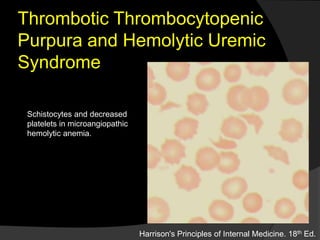


















Platelets are cell fragments that originate from megakaryocytes and play a key role in hemostasis. The normal platelet count is 150,000-450,000/L. Thrombopoietin regulates platelet production in the liver and spleen. When the endothelium is damaged, platelets adhere through von Willebrand factor and become activated, releasing granule contents that promote clot formation. Thrombocytopenia can result from decreased production, increased destruction, or sequestration and has various acquired and inherited causes. Disorders like immune thrombocytopenic purpura and thrombotic thrombocytopenic purpura involve autoimmune or inflammatory platelet destruction.











































































































