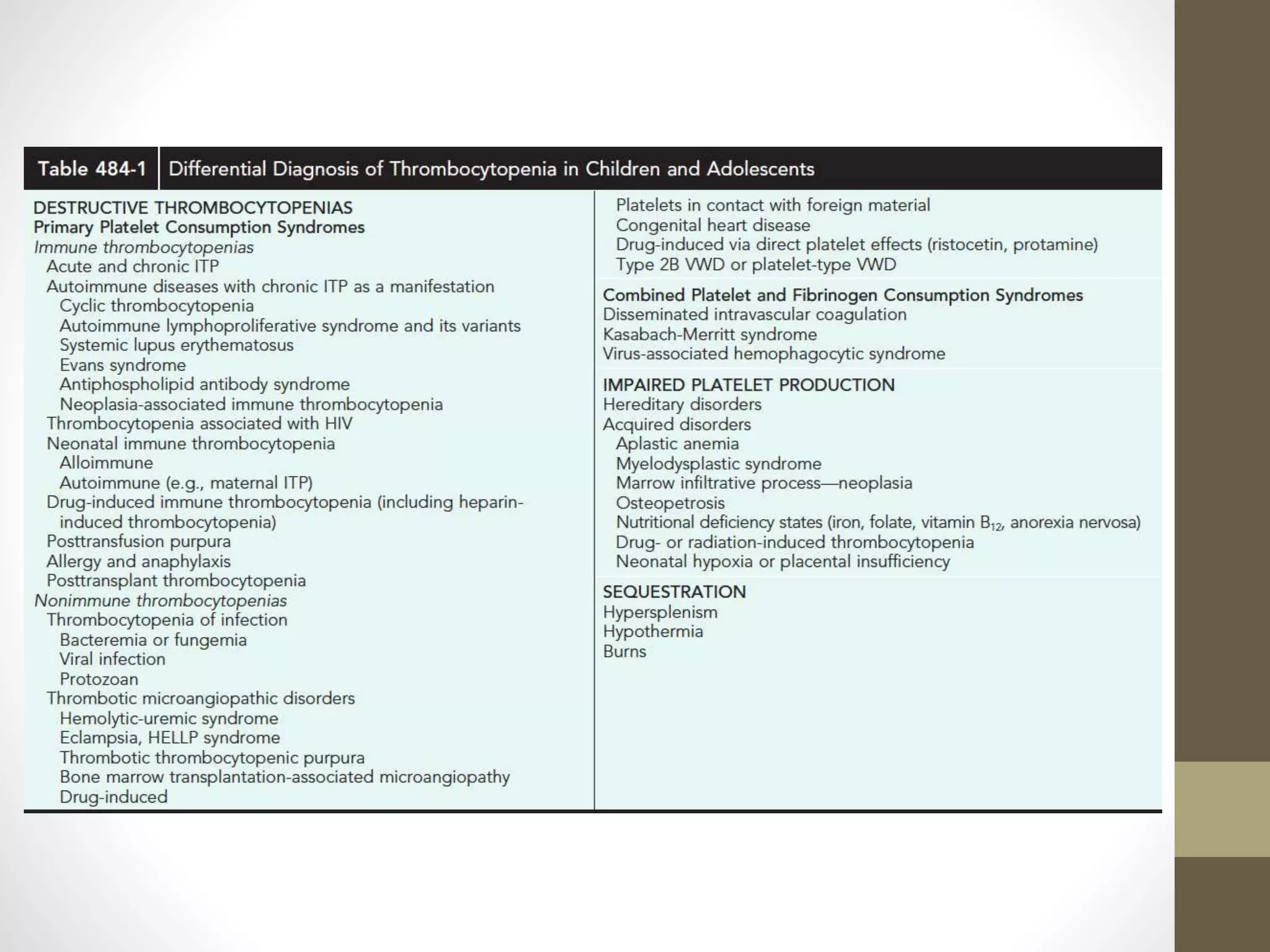
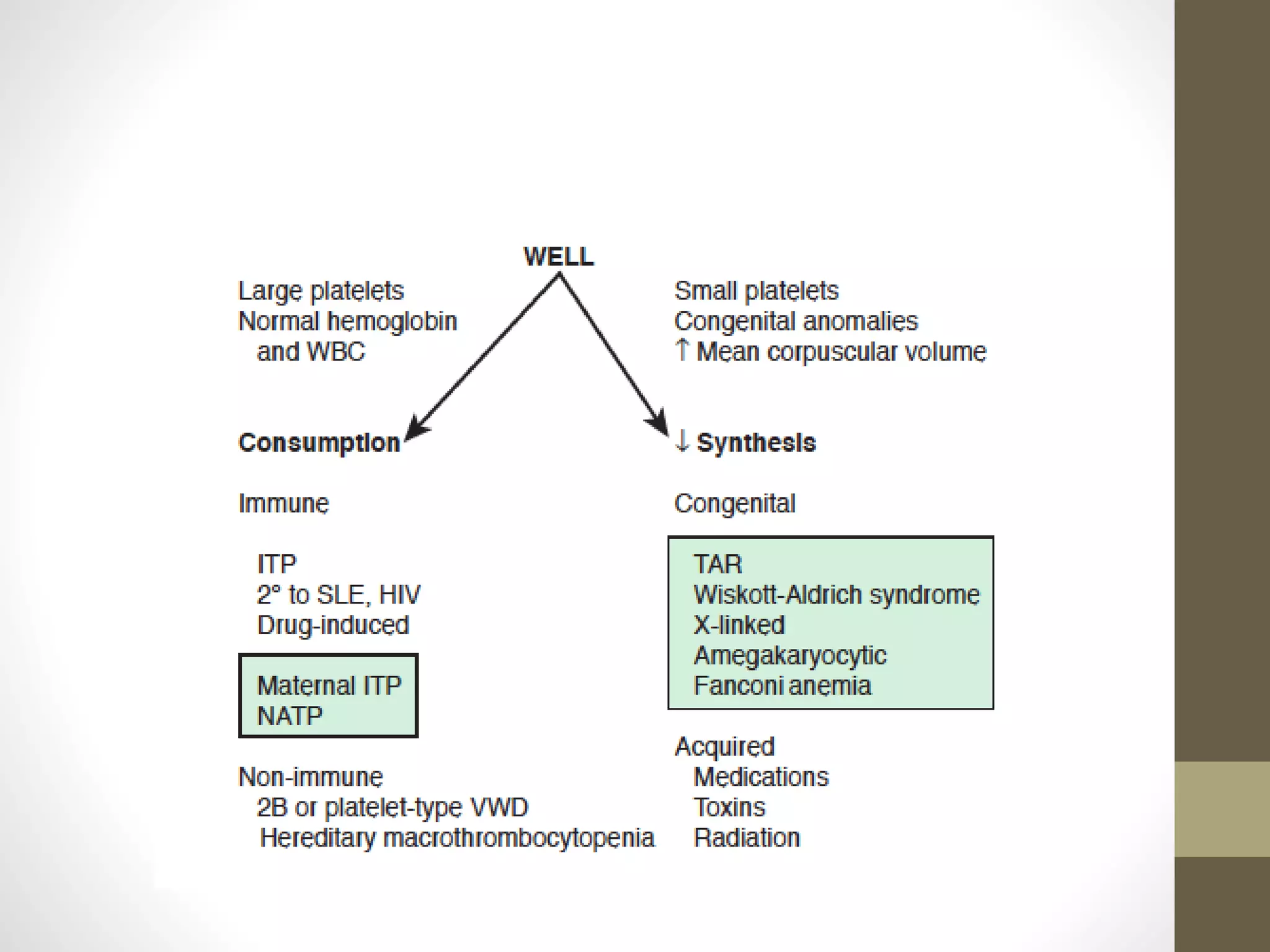
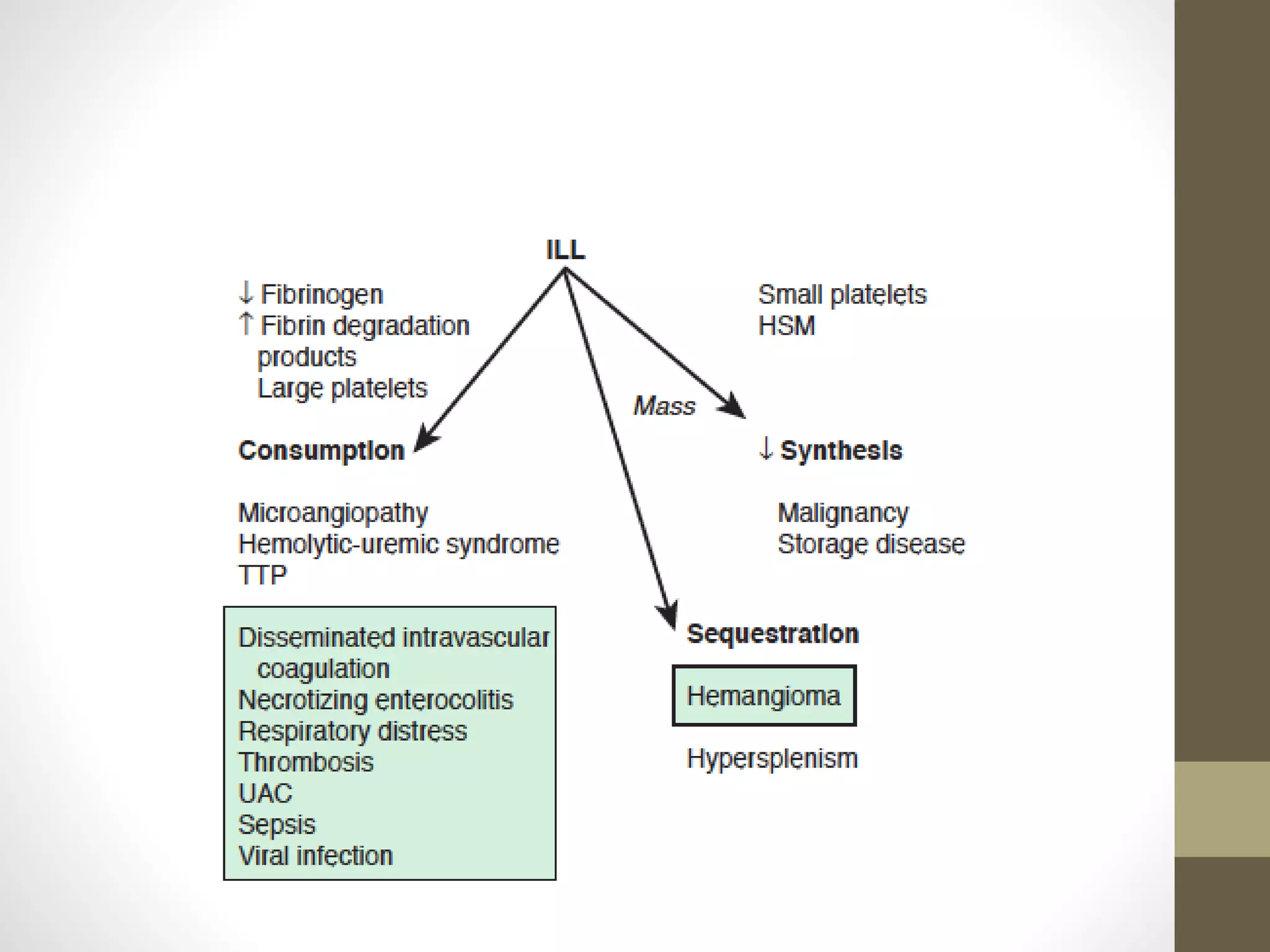
This document summarizes various platelet disorders including their causes, characteristics, diagnosis and treatment. It discusses decreased platelet production from bone marrow issues as well as increased platelet destruction from immune or non-immune causes. Specific disorders covered include idiopathic thrombocytopenic purpura, hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, sequestration, Kasabach-Merritt syndrome, and others. Diagnostic tests and treatment approaches are provided for each condition.