Downloaded 94 times








![Contents of alpha-granules con..
proteins that are either synthesized in megakaryocytes:
platelet factor 4 [PF4], β-thromboglobulin [βTG], VWF, platelet-derived
growth factor [PDGF]
endocytosed from plasma:
fibrinogen, albumin, immunoglobulin](https://image.slidesharecdn.com/plateletstoragepooldisorders-150705025011-lva1-app6891/85/Platelet-storage-pool-disorders-9-320.jpg)





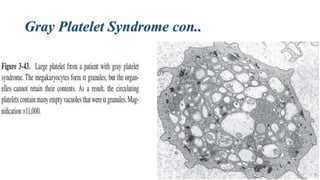






















Platelet storage pool disease (SPD) encompasses various disorders characterized by deficiencies in platelet granules, particularly alpha granules (α-granules) and dense granules (δ-granules). Key disorders include gray platelet syndrome, which results in poorly formed α-granules leading to bleeding issues, and Quebec platelet disorder, where delayed bleeding occurs due to abnormalities in α-granule protein degradation. Additionally, δ-granule disorders exhibit impaired platelet aggregation and have been associated with several syndromes affecting platelet function and survival.























































































