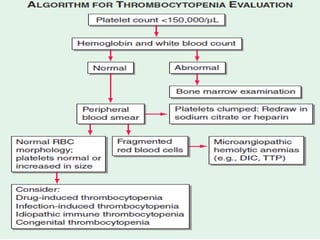
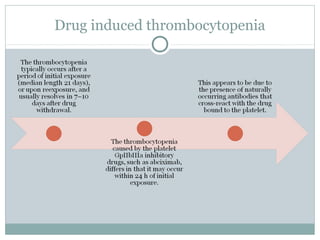
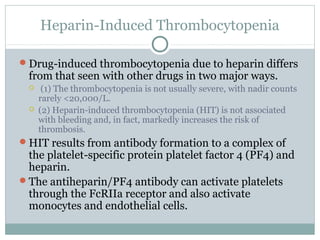
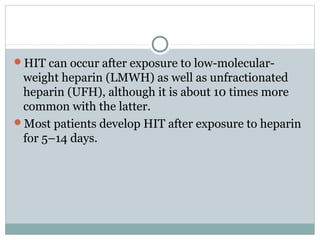
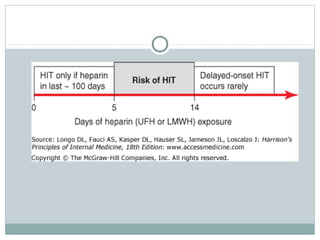
Platelets play a key role in hemostasis. Disorders of platelets can be quantitative, involving too few or too many platelets, or qualitative, involving platelet function defects. Common causes of thrombocytopenia include decreased production, increased destruction, sequestration, or pseudothrombocytopenia. Immune thrombocytopenic purpura is an immune-mediated disorder treated with steroids, IVIG, or splenectomy. Thrombotic thrombocytopenic purpura involves a deficiency in ADAMTS13 treated with plasma exchange. Von Willebrand disease involves defects in von Willebrand factor.



























































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)













