




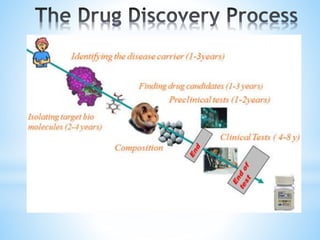
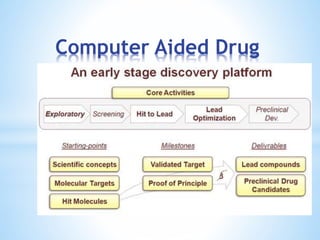

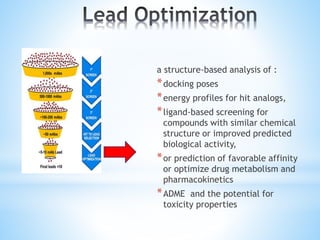
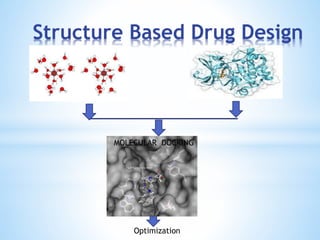


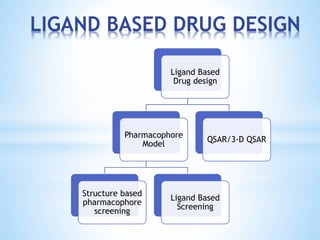

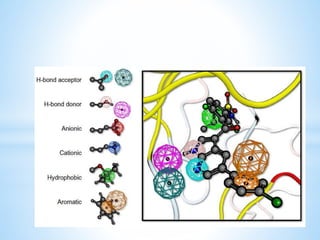
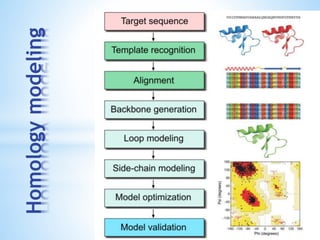

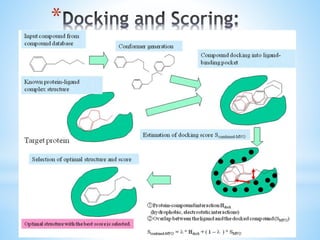
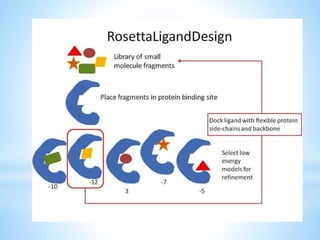
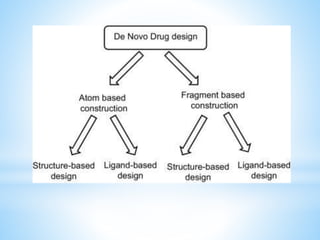
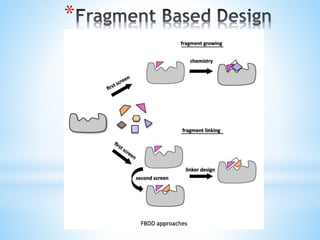
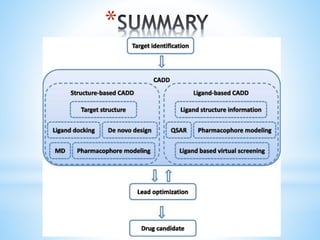



Computer aided drug design uses computational methods to aid in the drug discovery process. It can be used for both structure based drug design where the biological target structure is known, as well as ligand based drug design where the target structure is unknown. Structure based drug design techniques include molecular docking to study ligand binding poses and interactions. Ligand based techniques include pharmacophore modeling to identify chemical features important for activity and quantitative structure-activity relationships to correlate chemical structure to biological activity. These computational methods allow for more rapid and cost-effective discovery and optimization of drug candidates compared to traditional experimental methods alone.























































































