Downloaded 26 times







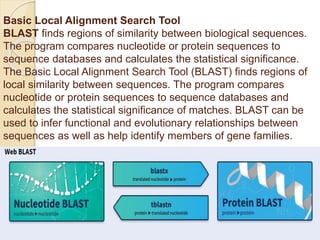

















This presentation introduces BLAST (Basic Local Alignment Search Tool) which is used to compare gene and protein sequences against databases. It discusses the types of BLAST programs including blastn, blastp, and PSI-BLAST. The BLAST algorithm works by removing repeats, making a word list from the query, searching the database for matches, and extending matches. BLAST output includes alignments and statistical values. Key functions of BLAST are locating domains, establishing phylogeny, DNA mapping, and comparison. The objectives are to enable comparison of a query to database sequences and identify related matches above a threshold.























































































