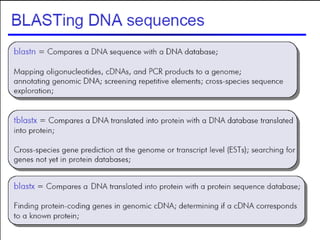
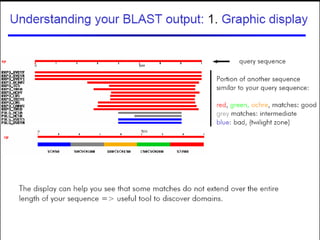
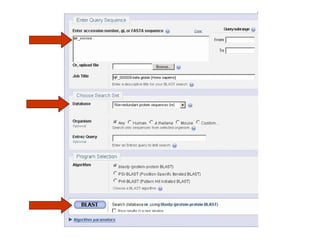
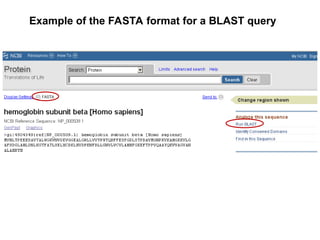
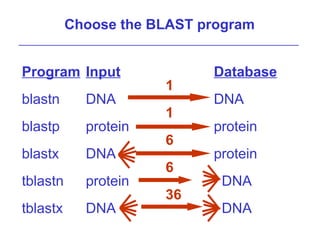
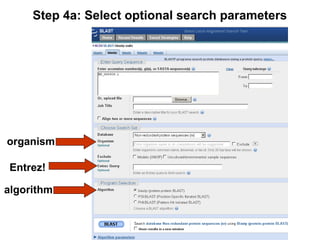
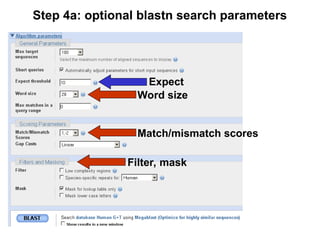
BLAST (Basic Local Alignment Search Tool) is a widely used bioinformatics algorithm for comparing biological sequences against databases to identify statistically significant similarities. It operates using a heuristic method that focuses on local alignments and features three main components: seeding, extension, and evaluation. Its applications include identifying unknown sequences, phylogenetic analysis, and exploring protein structure and function, with various adjustable parameters for searching different types of sequence data.




























































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)













