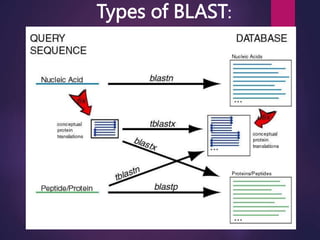
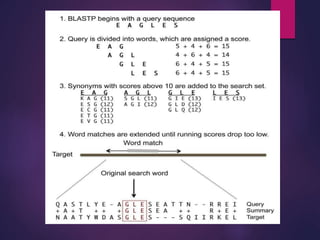
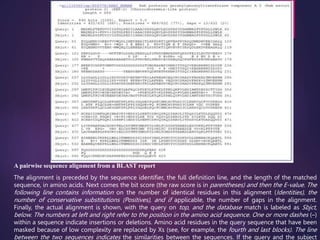
BLAST (Basic Local Alignment Search Tool), developed by Steven Altschul and Samuel Karlin in 1990, is a heuristic method used to compare nucleotide and amino acid sequences for similarities. It operates by breaking down a query sequence into short overlapping words and assesses matches using scoring matrices, with various types such as blastp and blastn tailored for different sequence comparisons. The results provide alignments ranked by score and statistical significance, facilitating the identification of similar sequences and their potential functions.