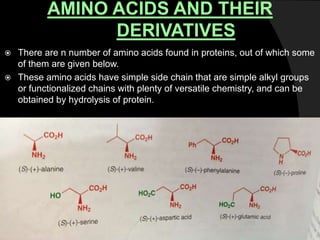
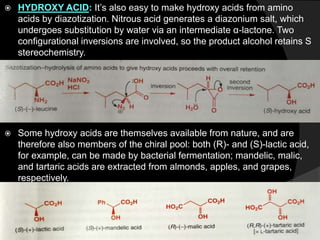
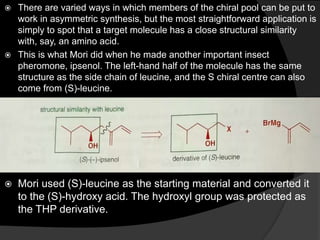
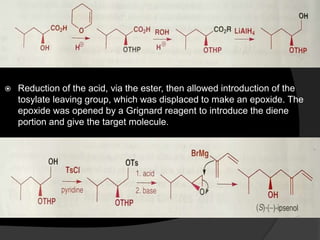
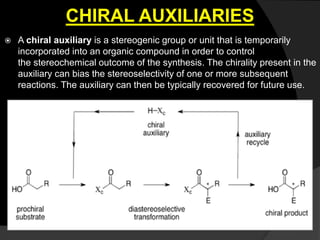
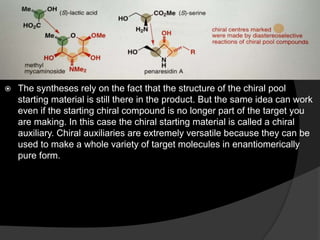
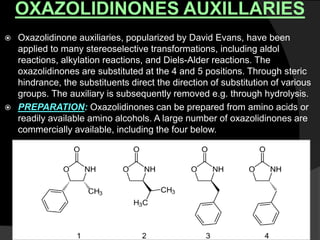
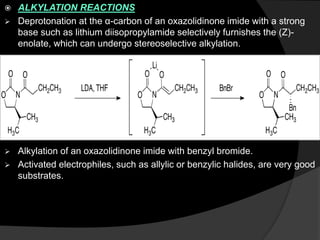
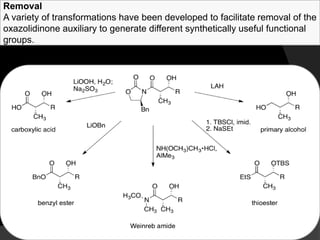
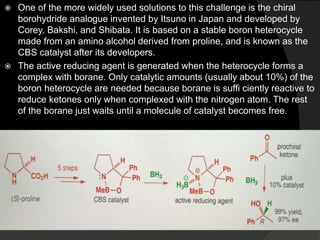
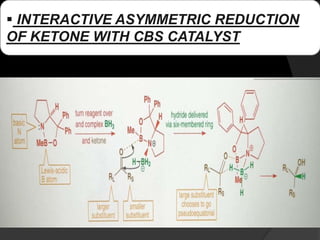
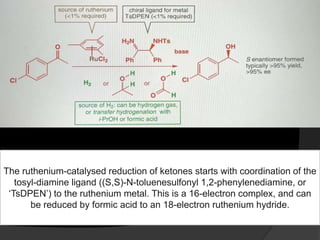
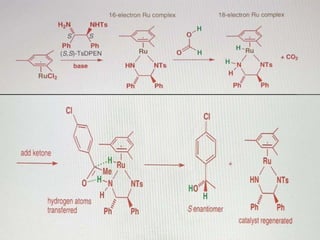
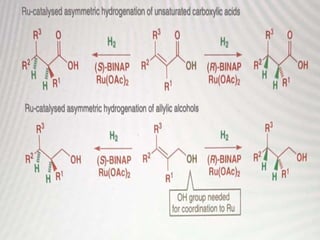
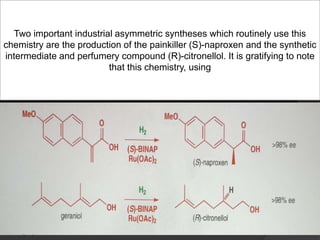
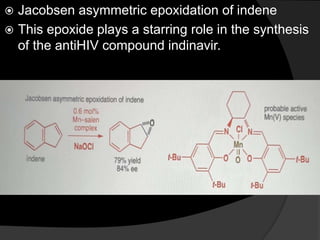
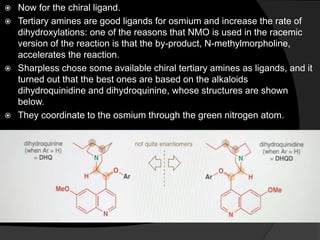
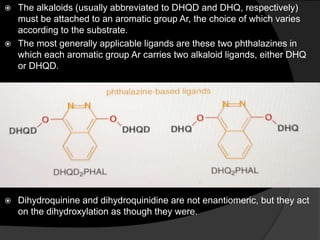
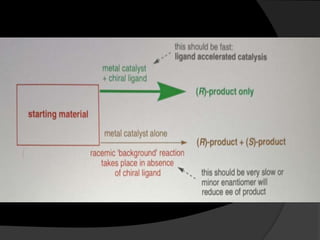
This document discusses various methods for asymmetric synthesis, which is a form of chemical synthesis that favors the formation of one stereoisomer over another. It begins by explaining enantioselective synthesis and its importance in pharmaceuticals. It then discusses using naturally occurring chiral compounds as starting materials, known as the "chiral pool". Examples of compounds in the chiral pool are discussed, such as amino acids and carbohydrates. Methods for using these compounds or derivatives in asymmetric synthesis are provided, such as through diastereoselective reactions. The document also discusses using chiral auxiliaries and catalysts to control stereoselectivity in reactions. Specific examples of chiral auxiliaries like oxazolidinones and catalytic reactions like asymmetric