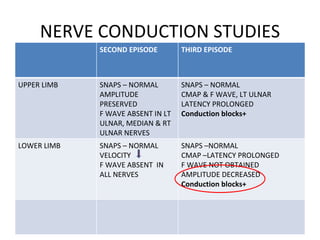
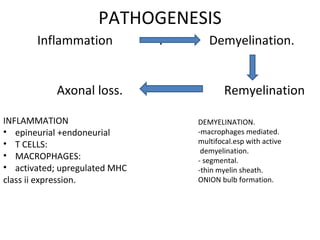
This document describes a case of a 32-year-old male who presented with weakness in all four limbs that progressed over a week. Based on examination and investigations, he was diagnosed with recurrent quadriparesis likely due to an autoimmune etiology of chronic inflammatory demyelinating polyneuropathy (CIDP) associated with systemic lupus erythematosus (SLE). He showed improvement in symptoms with intravenous steroids and plasma exchange.









































![Despite these limitations, early diagnosis and treatment is important in preventing irreversible axonal loss and improving functional recovery. [2] Lack of awareness and treatment of CIDP is also due to limitations of clinical trials. Although there are stringent research criteria for selecting patients to clinical trials, Application of the present research criteria to routine clinical practice often miss the diagnosis in a majority of patients, and patients are often left untreated despite progression of their disease. [3] In some cases electrophysiological studies fail to show any evidence of demyelination. Though conventional electrophysiological diagnostic criteria are not met, the patient may still respond to immunomodulatory treatments. In such cases, presence of clinical characteristics suggestive of CIDP are critical, justifying full investigations, including sural nerve biopsy. [4]](https://image.slidesharecdn.com/presentation1-110701060510-phpapp01/85/A-Case-of-CIDP-43-320.jpg)
![. [5] IVIG and plasmapheresis have proven benefit in randomized, double-blind, placebo-controlled trials. Despite less definitive published evidence of efficacy, corticosteroids are considered standard therapies because of their long history of use and cost effectiveness. IVIG is probably the first-line CIDP treatment, but is extremely expensive (in the U.S., a single 65 g dose of Gamunex brand in 2010 might be billed at the rate of $8,000, just for the immunoglobulin, not including other charges such as nurse administration). Gamunex brand IVIG is the only U.S. FDA approved treatment for CIDP, as in 2008 Talecris, the maker of Gamunex, received orphan drug status for this drug for the treatment of CIDP. Immunosuppressive drugs are often of the cytotoxic (chemotherapy) class, including Rituximab (Rituxan) which targets B Cells , and cyclophosphamide, a drug which reduces the function of the immune system. Ciclosporin has also been used in CIDP but with less frequency as it is a newer approach. [6] Ciclosporin is thought to bind to immunocompetent lymphocytes, especially T-lymphocytes. Non-cytotoxic immunosuppressive treatments usually include the anti-rejection transplant drugs azathioprine (Imuran) and mycophenolate mofetil (Cellcept).](https://image.slidesharecdn.com/presentation1-110701060510-phpapp01/85/A-Case-of-CIDP-44-320.jpg)













































![2009 Convegno Malattie Rare Nobile Orazio [22 01]](https://cdn.slidesharecdn.com/ss_thumbnails/2009convegnomalattierarenobileorazio2201-090330150653-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)

















































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)

![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)












