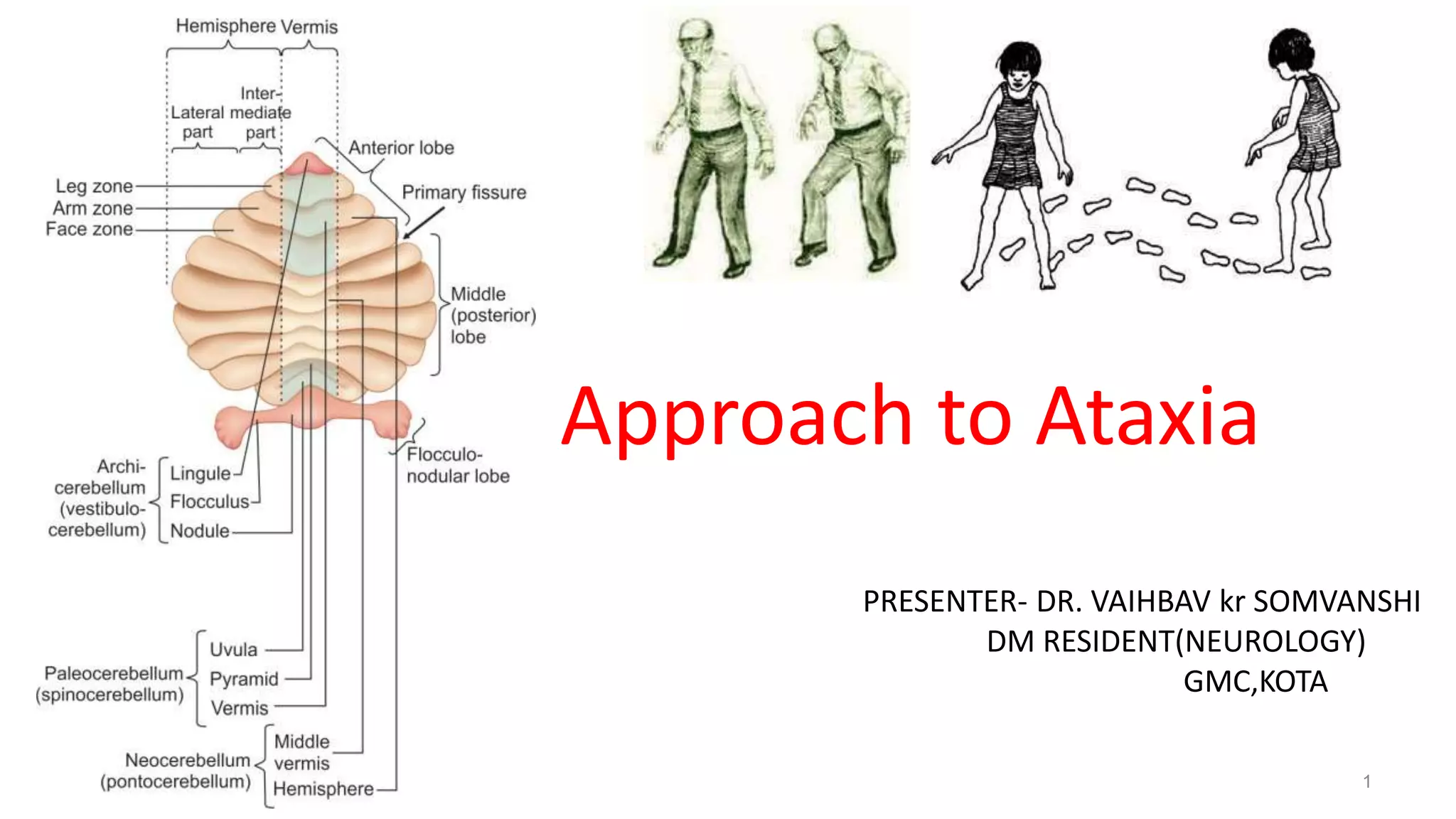
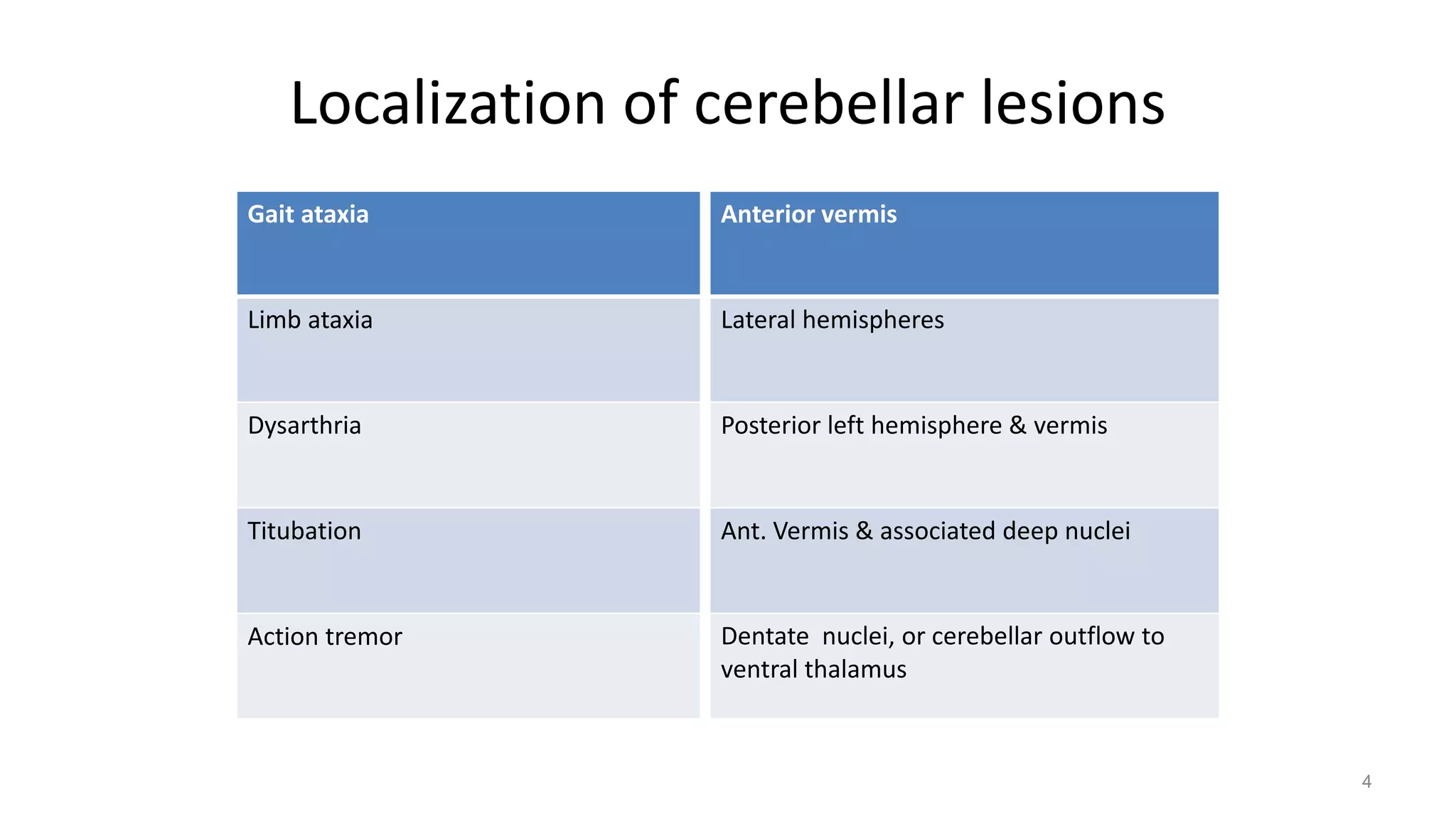
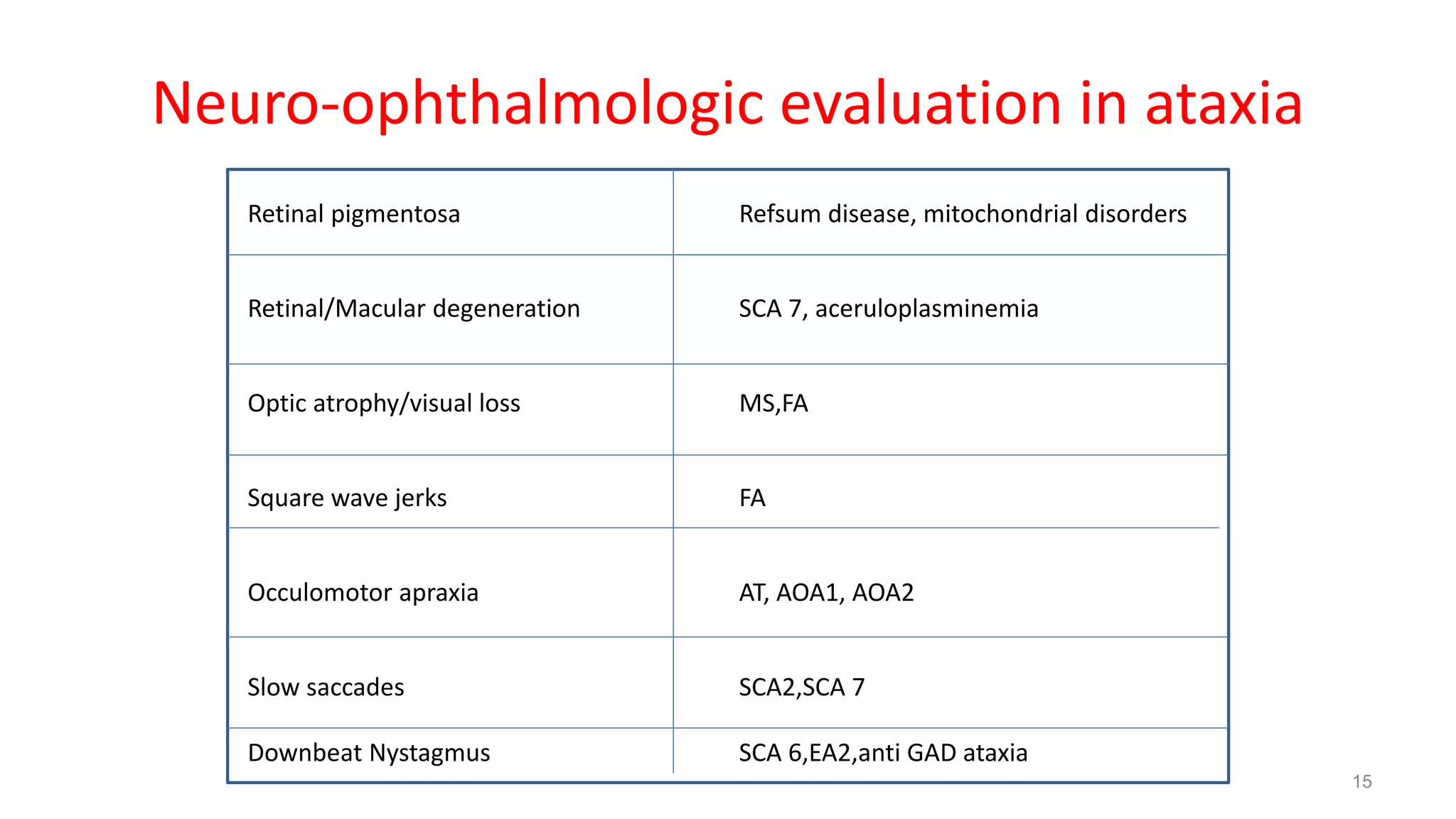
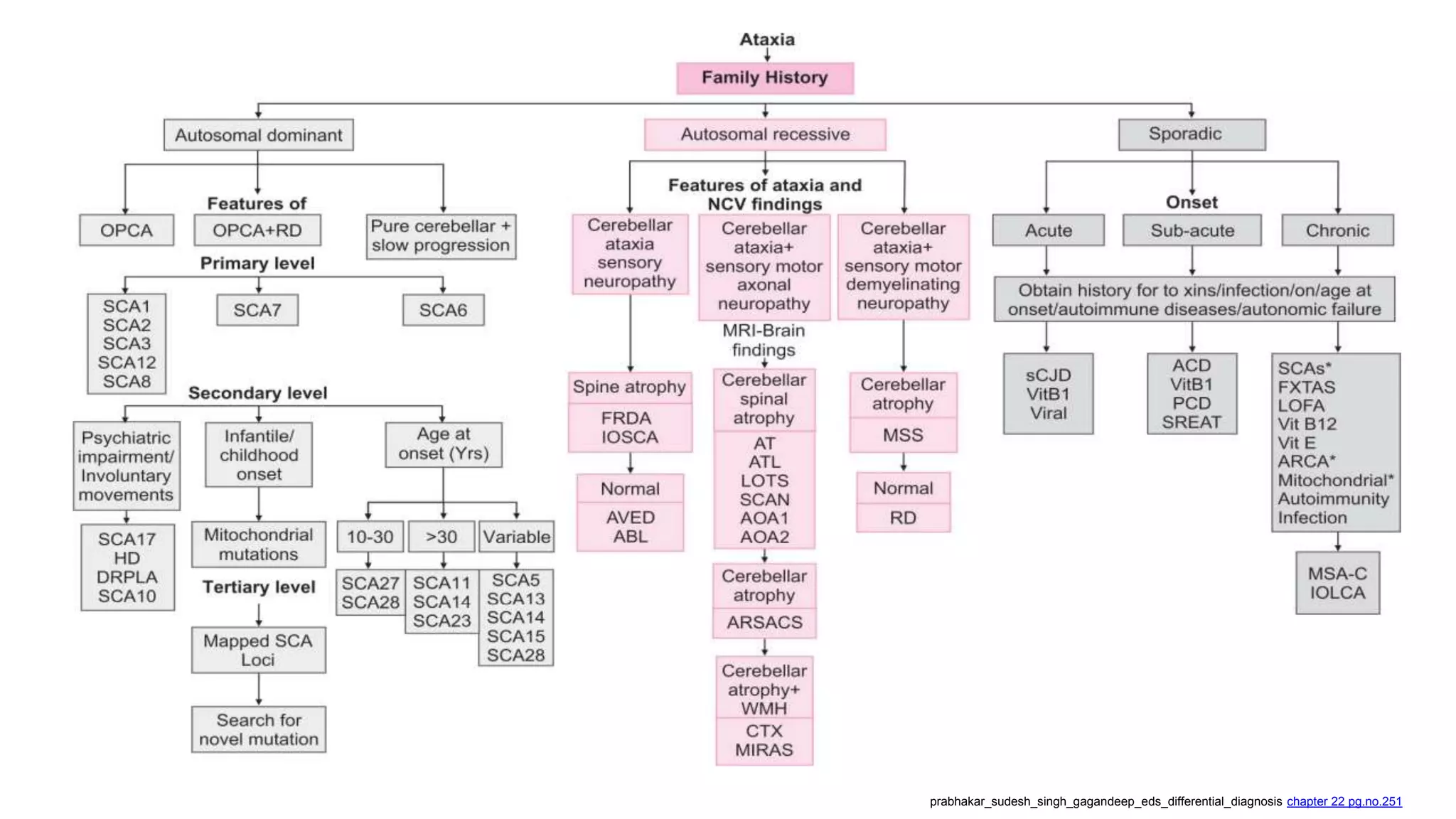
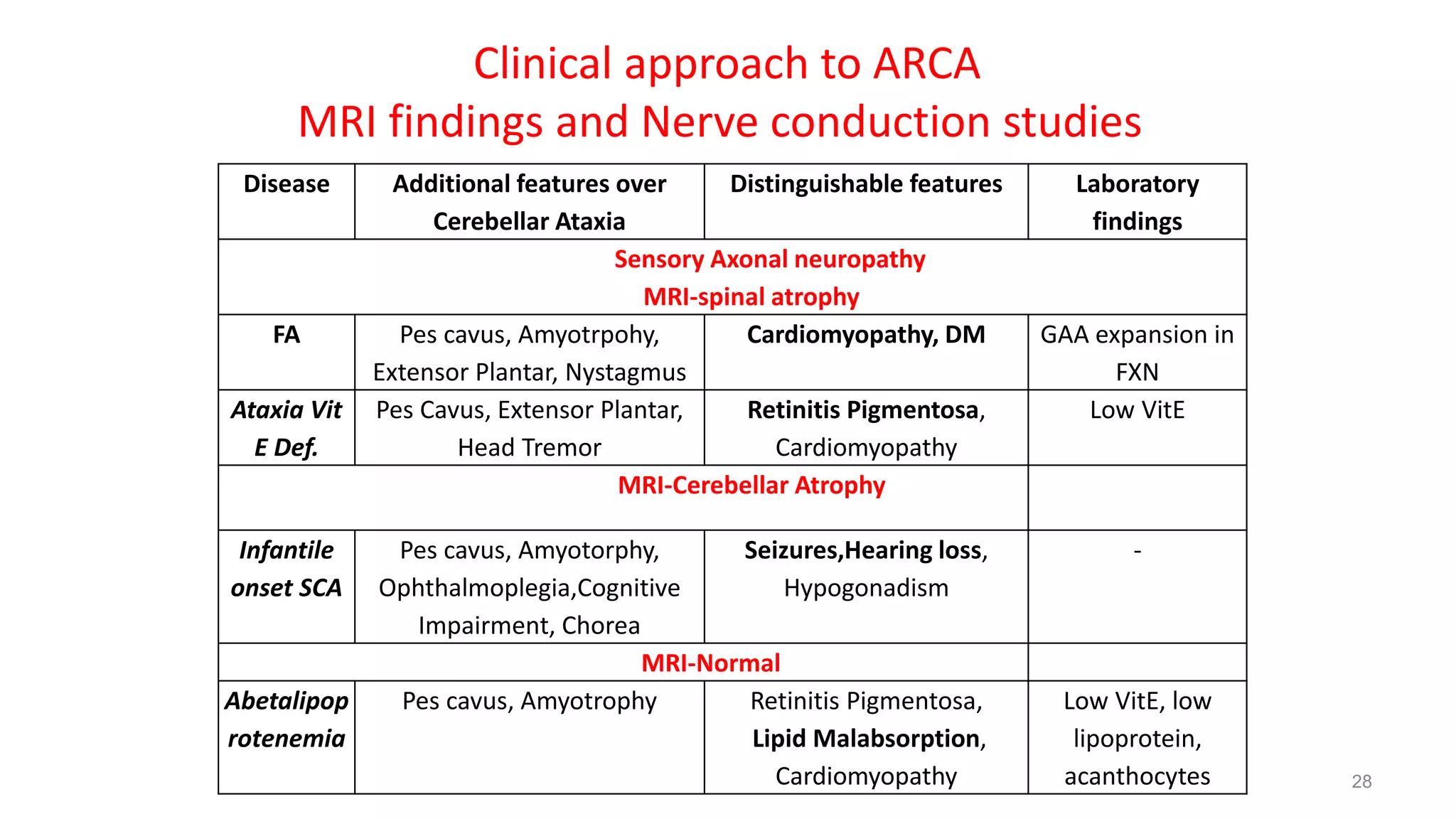
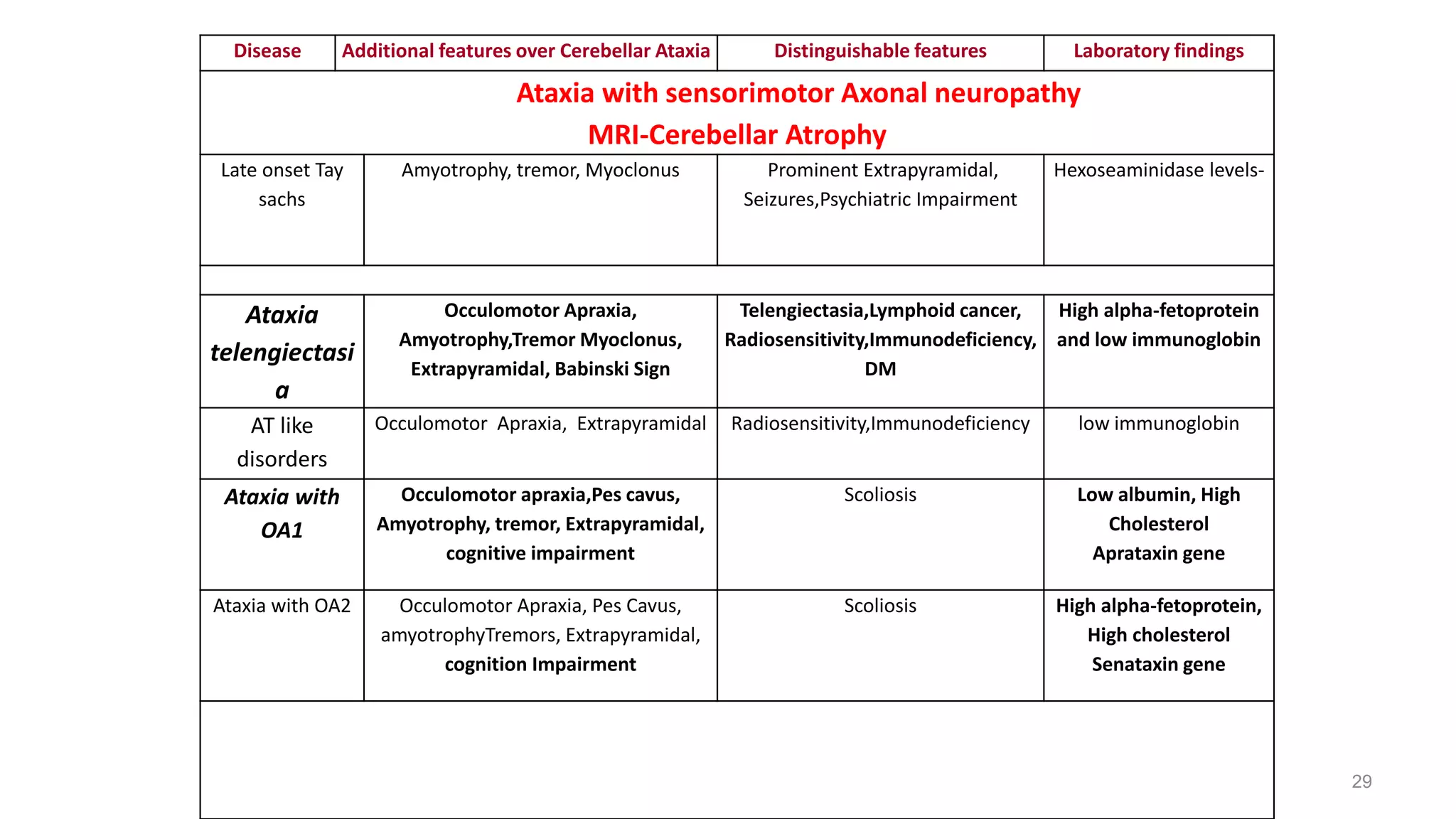
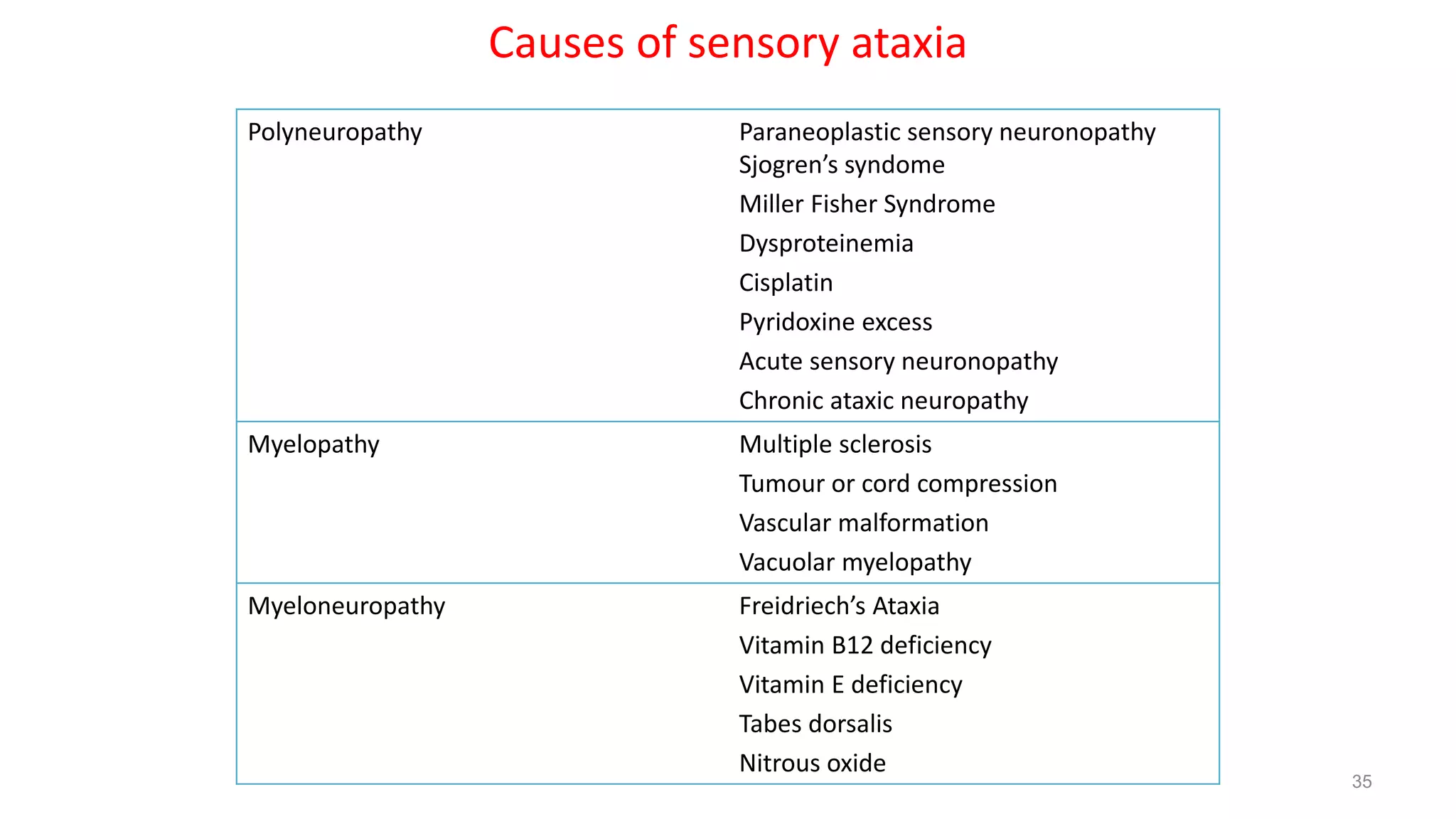
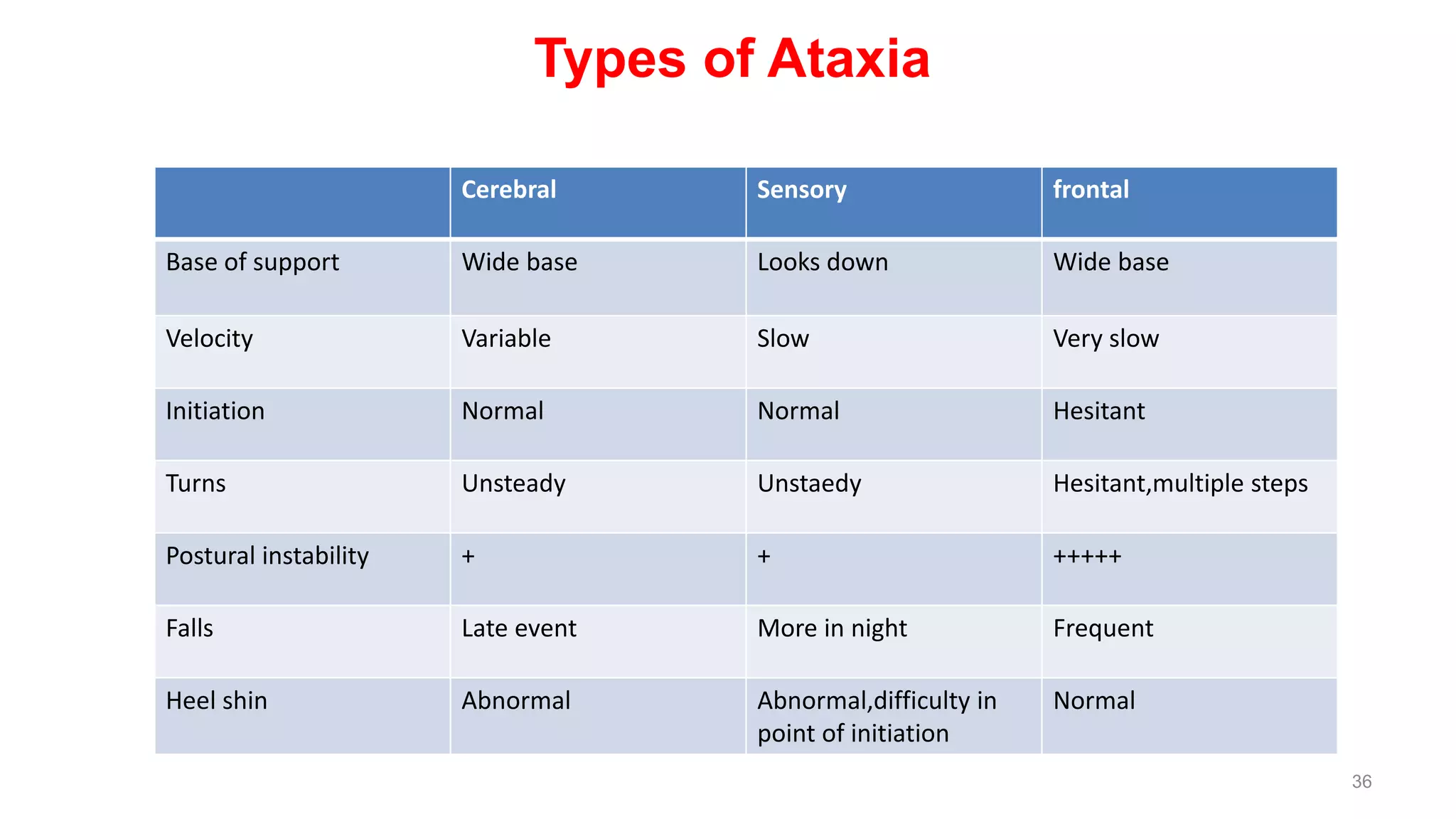
This document provides an overview of the approach to patients presenting with ataxia. It discusses the localization and causes of ataxia based on the involved neurological structures like the cerebellum and sensory pathways. Specific signs help to localize lesions within the cerebellum. A thorough history and examination along with targeted investigations can help identify acquired, genetic and other causes of ataxia. Neuroimaging, electrodiagnostic tests, ophthalmological and genetic testing are important to classify the type and guide management of ataxia.
![Introduction
• Ataxia = from Greek- a- [lack of]+ taxia [order]
• Rate, rhythm and force of contraction of voluntary movements
• Disorganized, poorly coordinated, or clumsy movements
Traditionally used specifically for lesions involving
• Cerebellum or it’s pathways
• Proprioceptive sensory pathways](https://image.slidesharecdn.com/approachtoataxia-210914051954/75/Approach-to-Ataxia-2-2048.jpg)






























































































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)






























