Downloaded 388 times

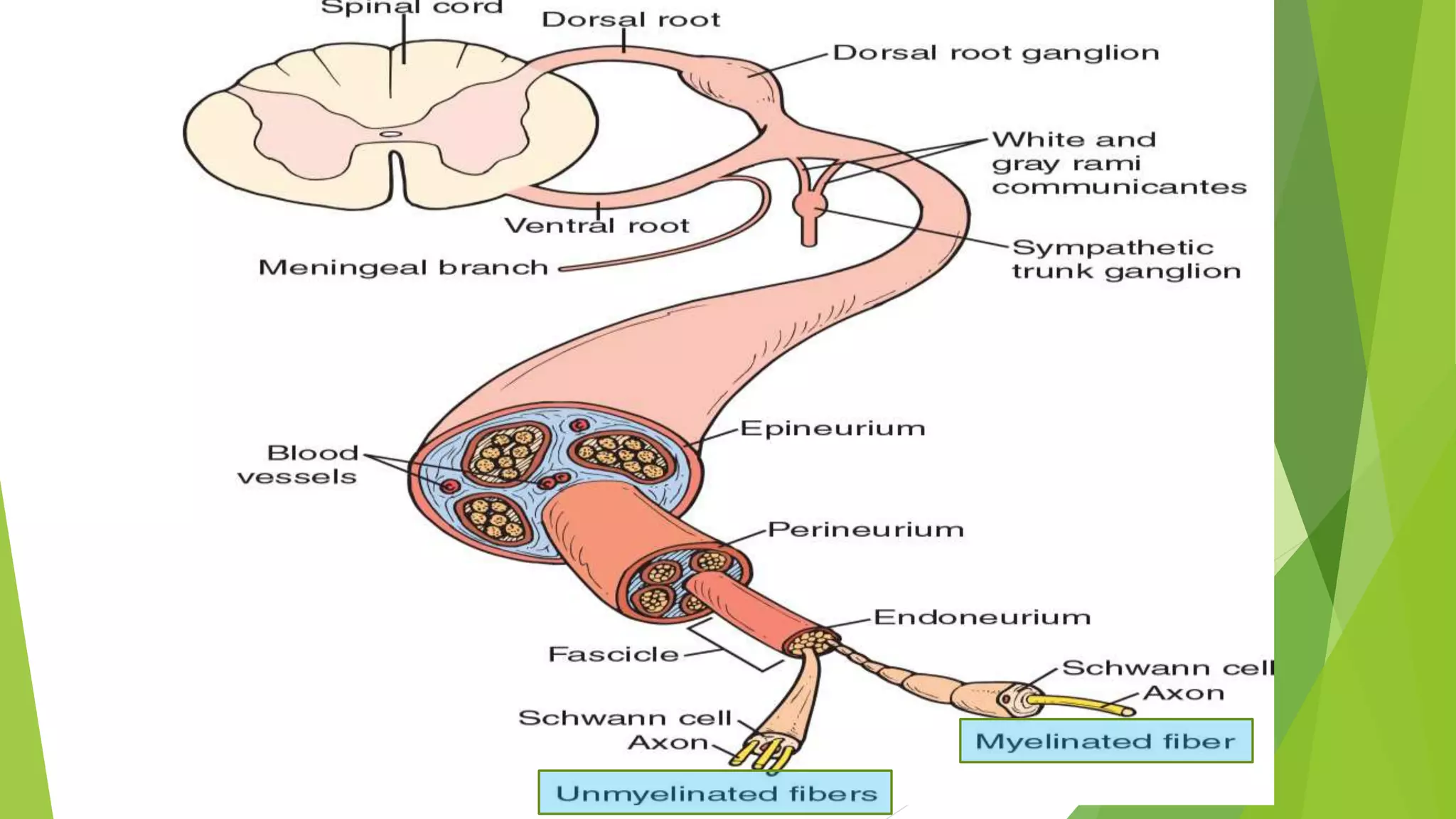
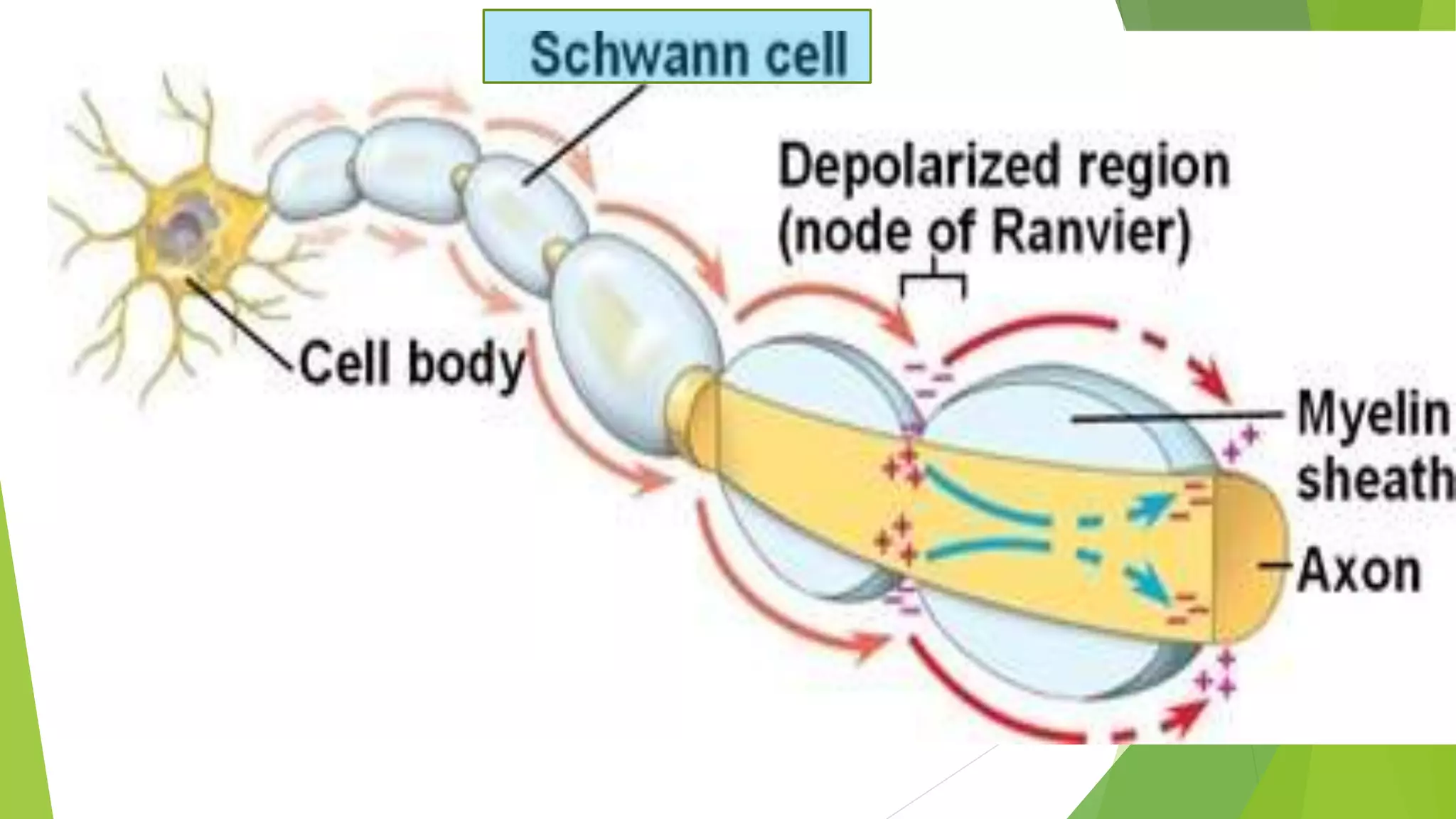

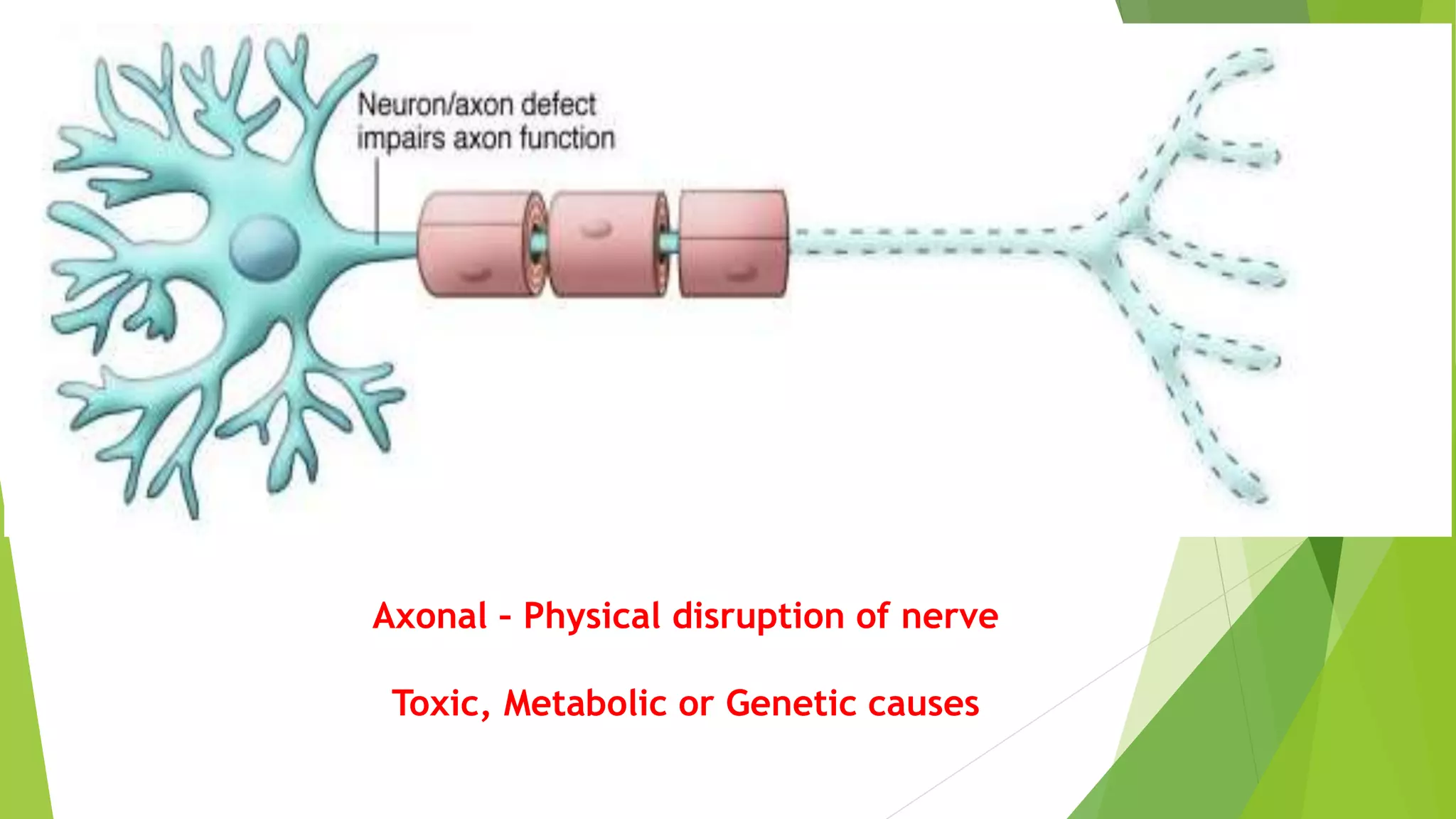

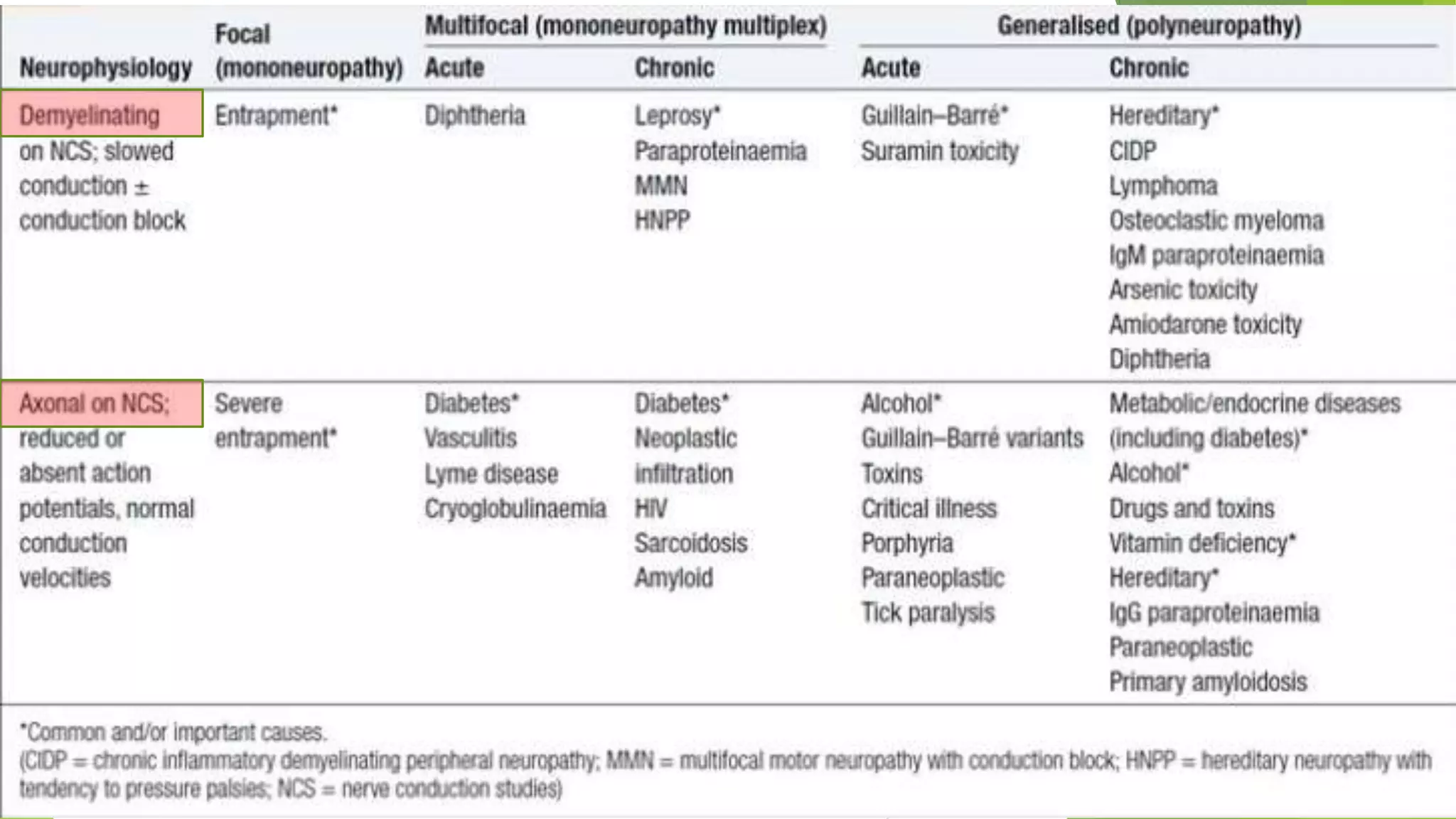










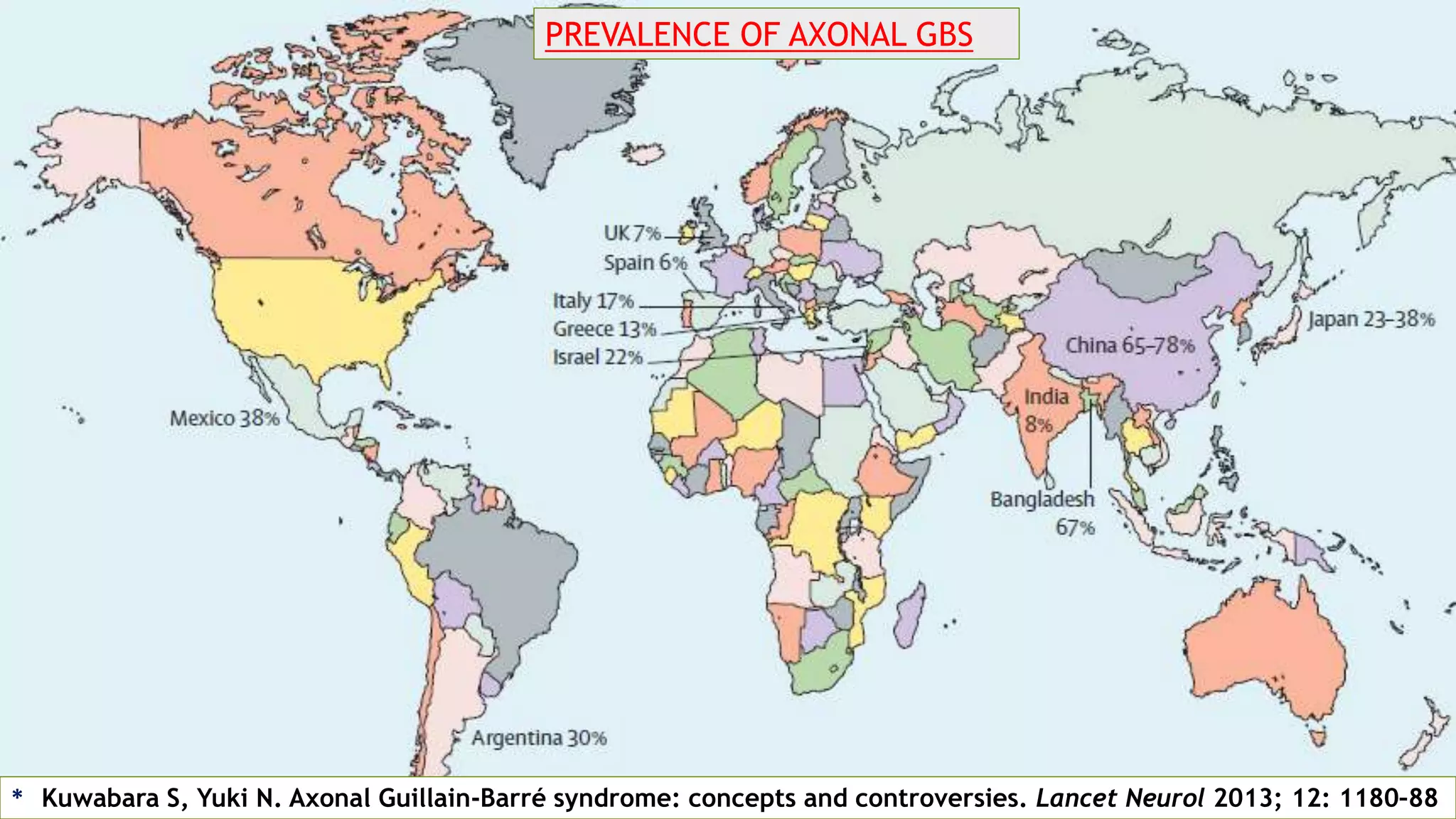







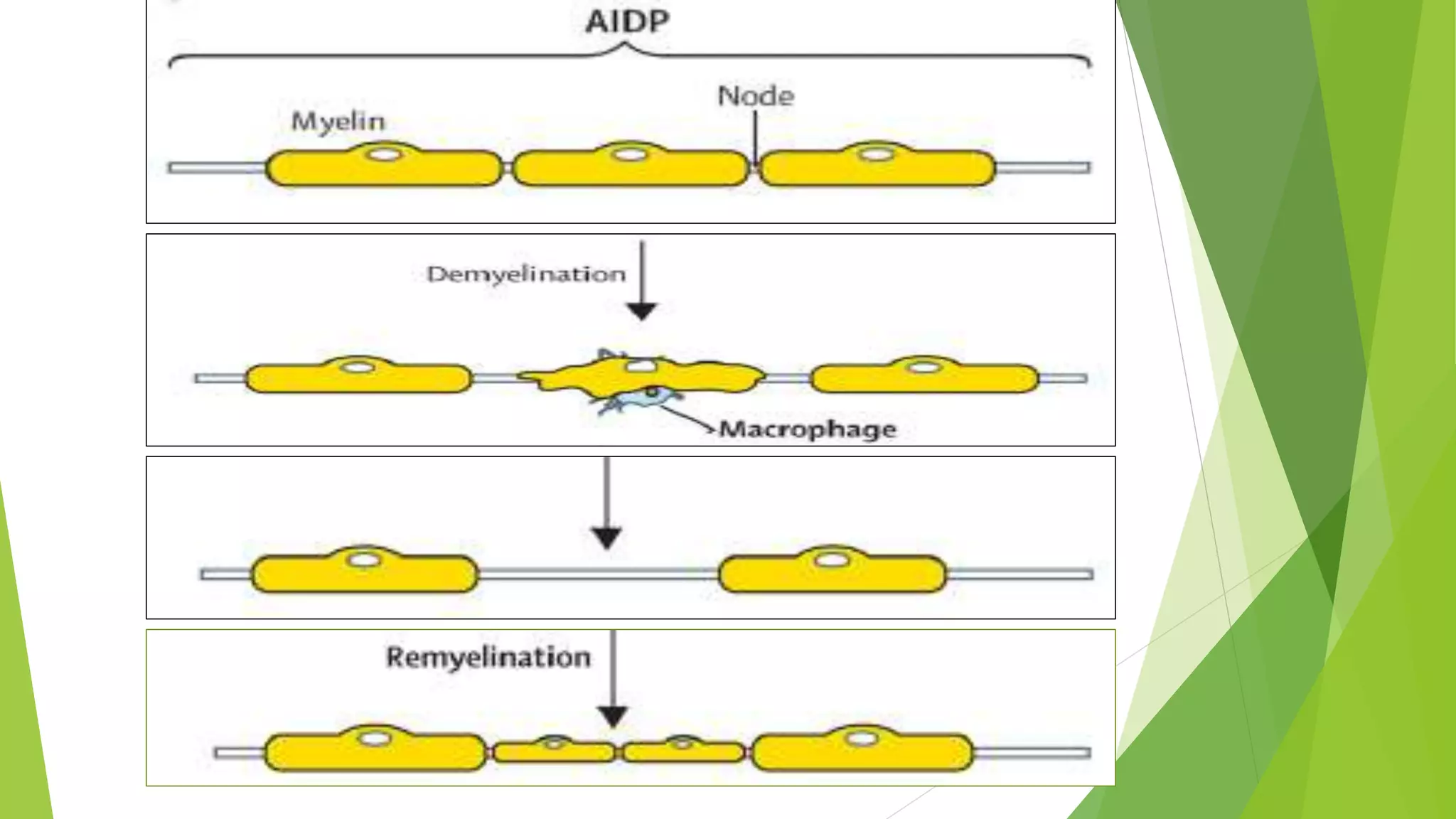




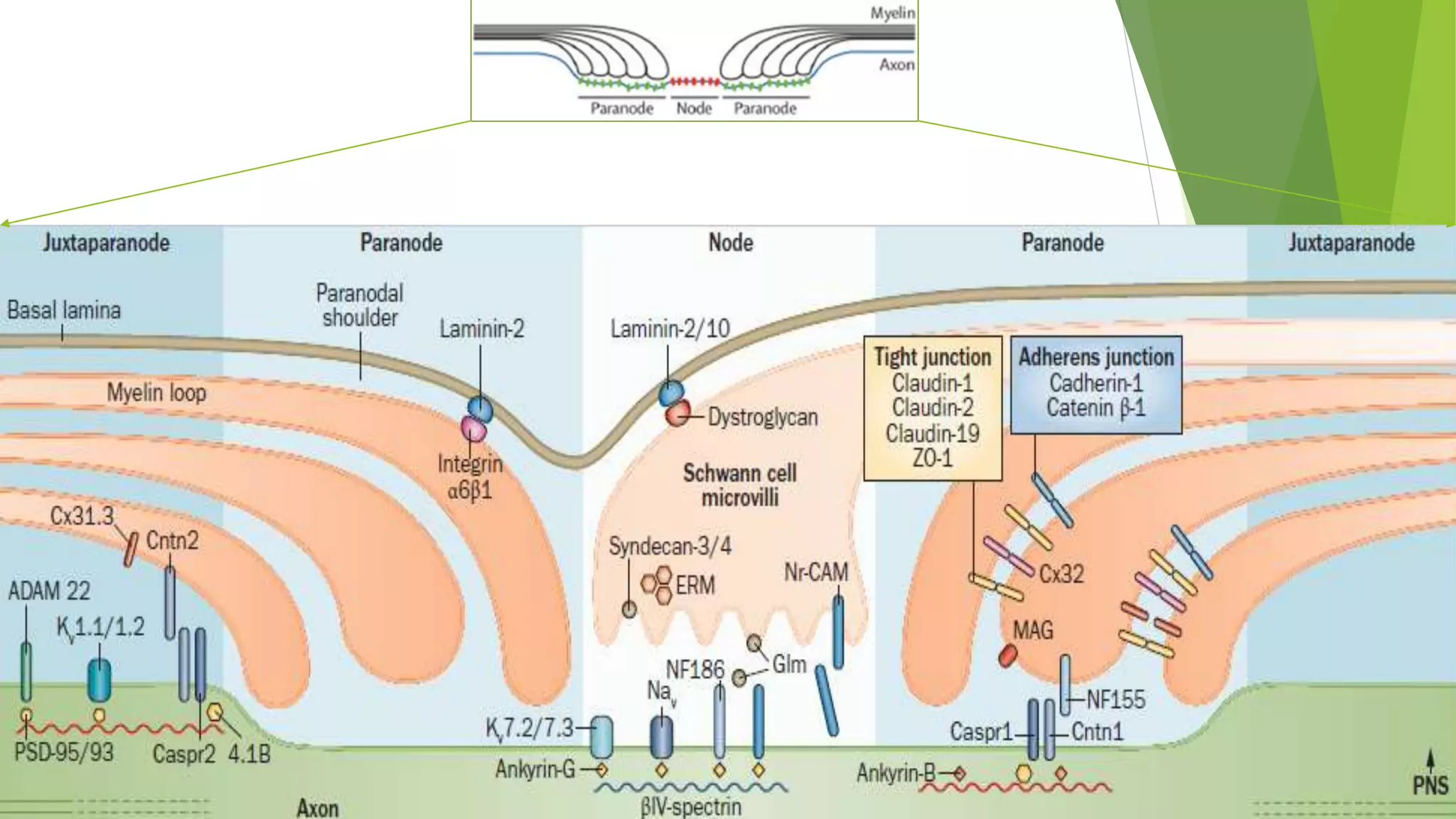

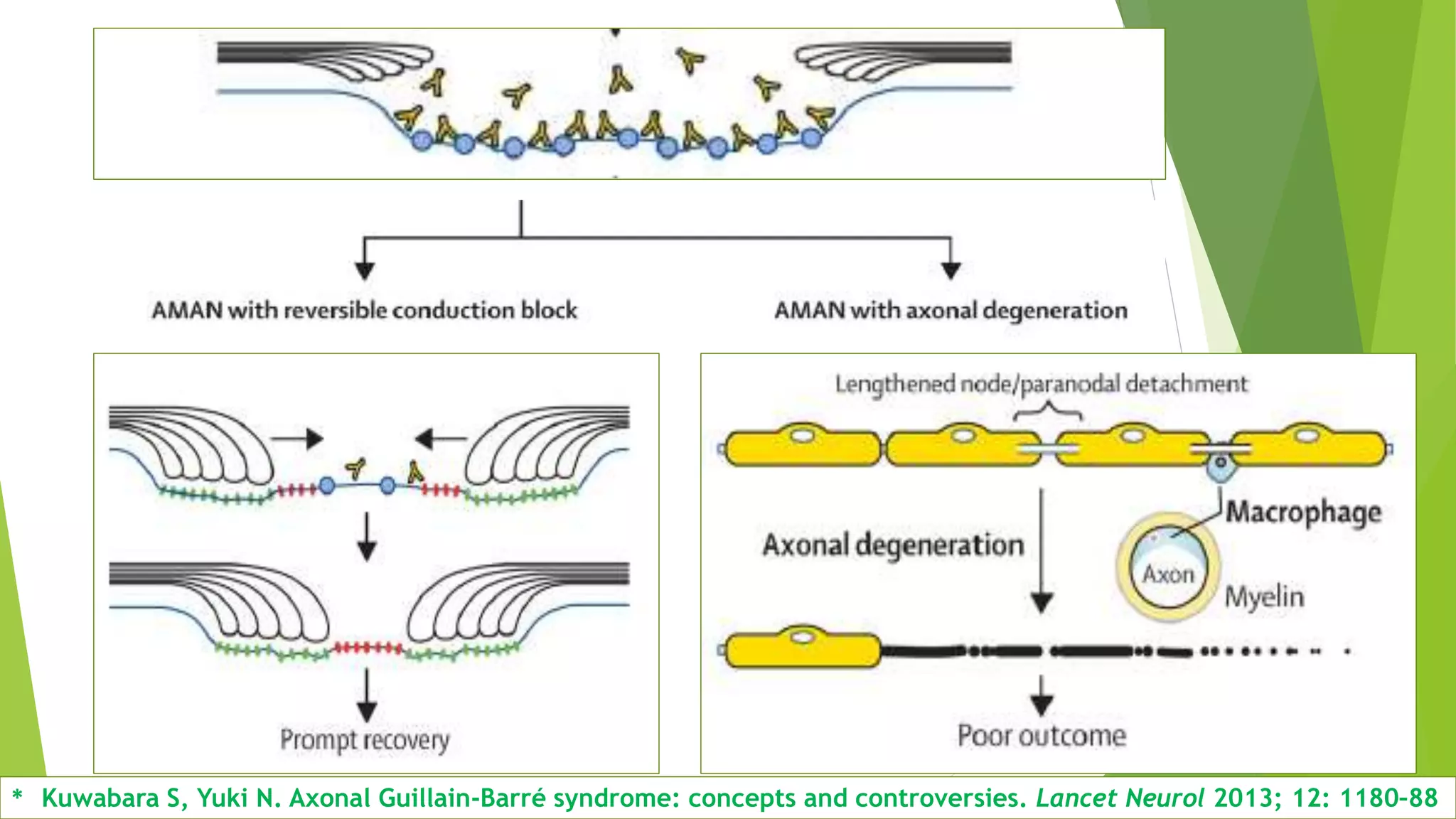










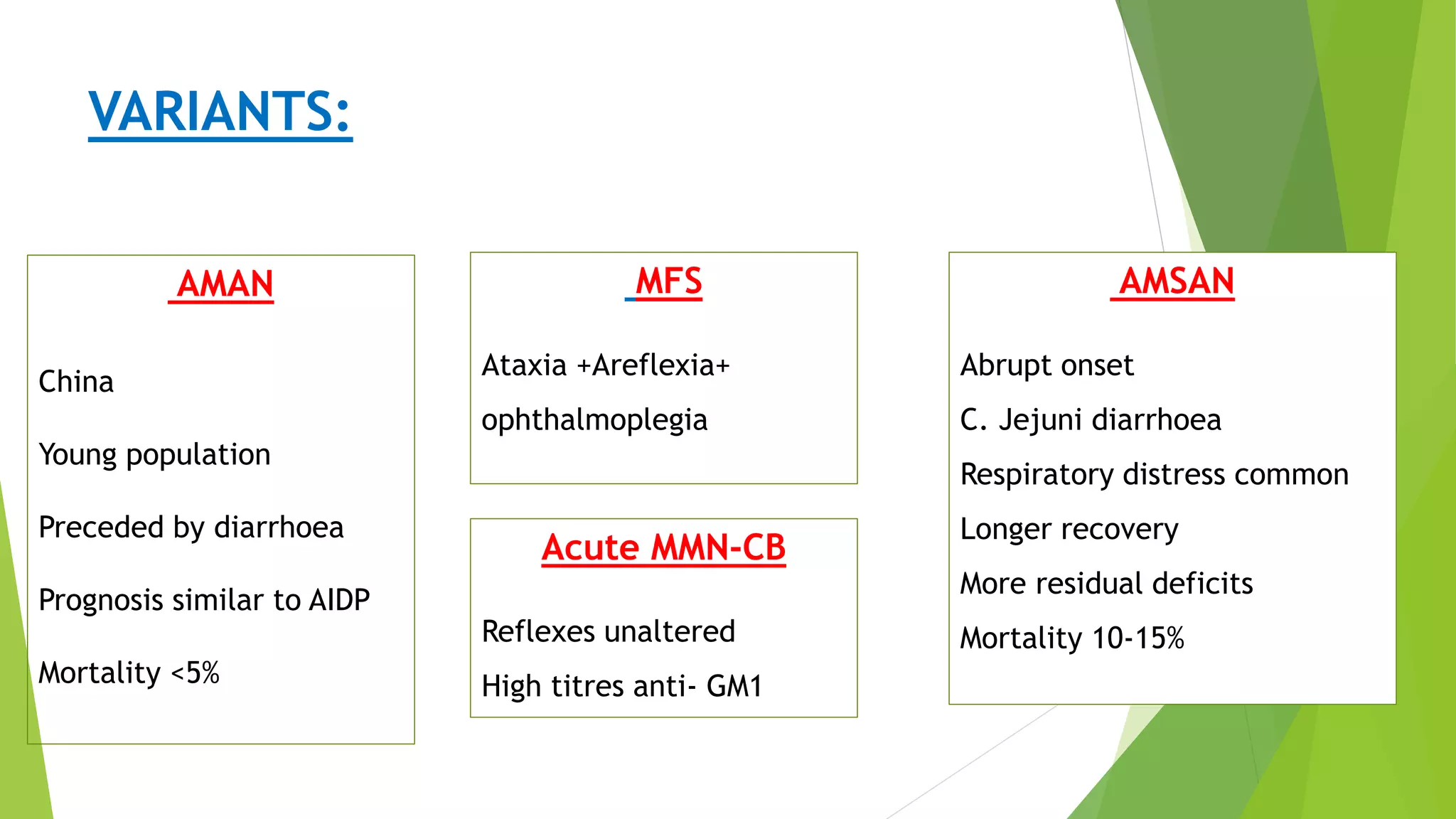
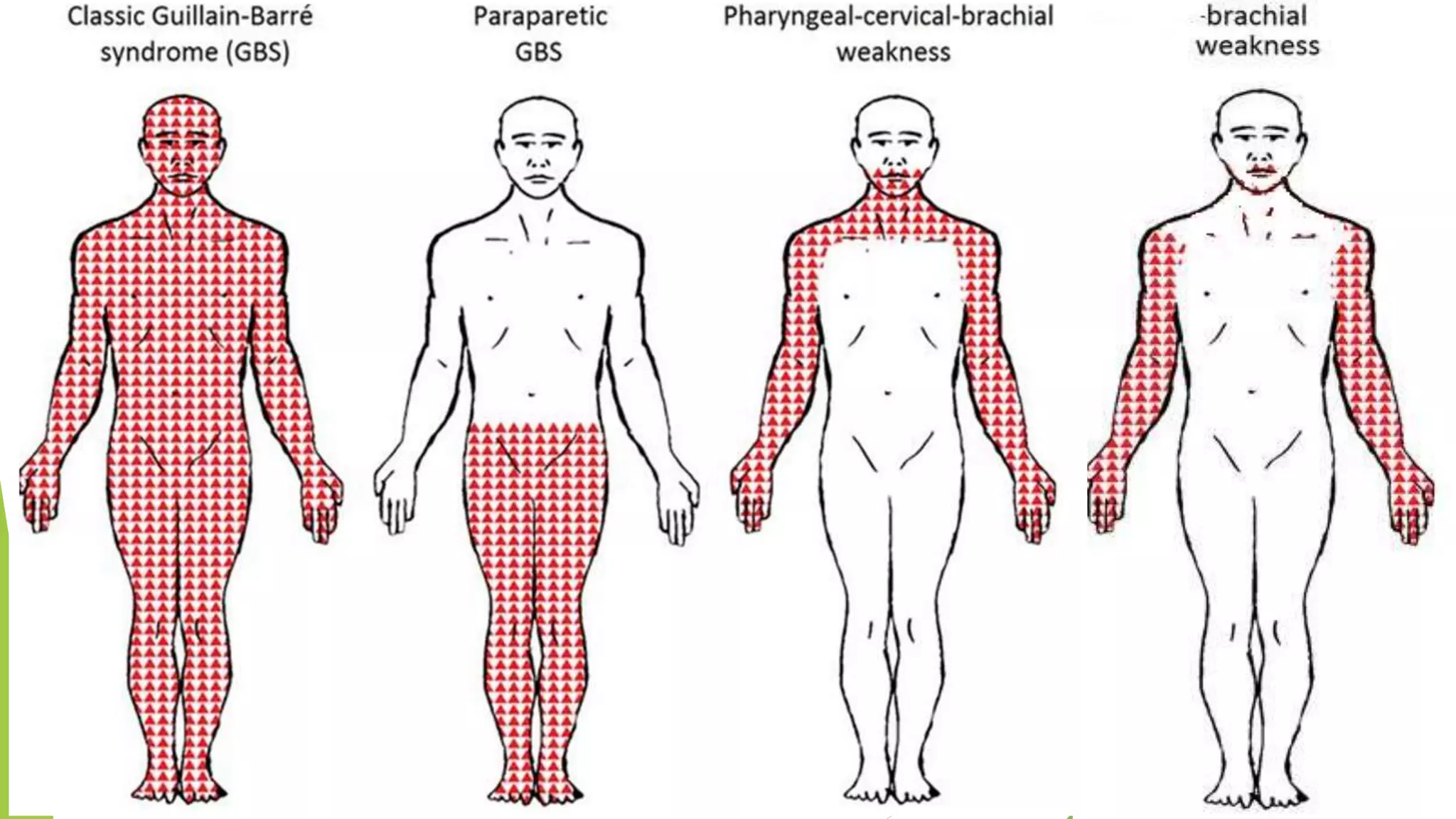
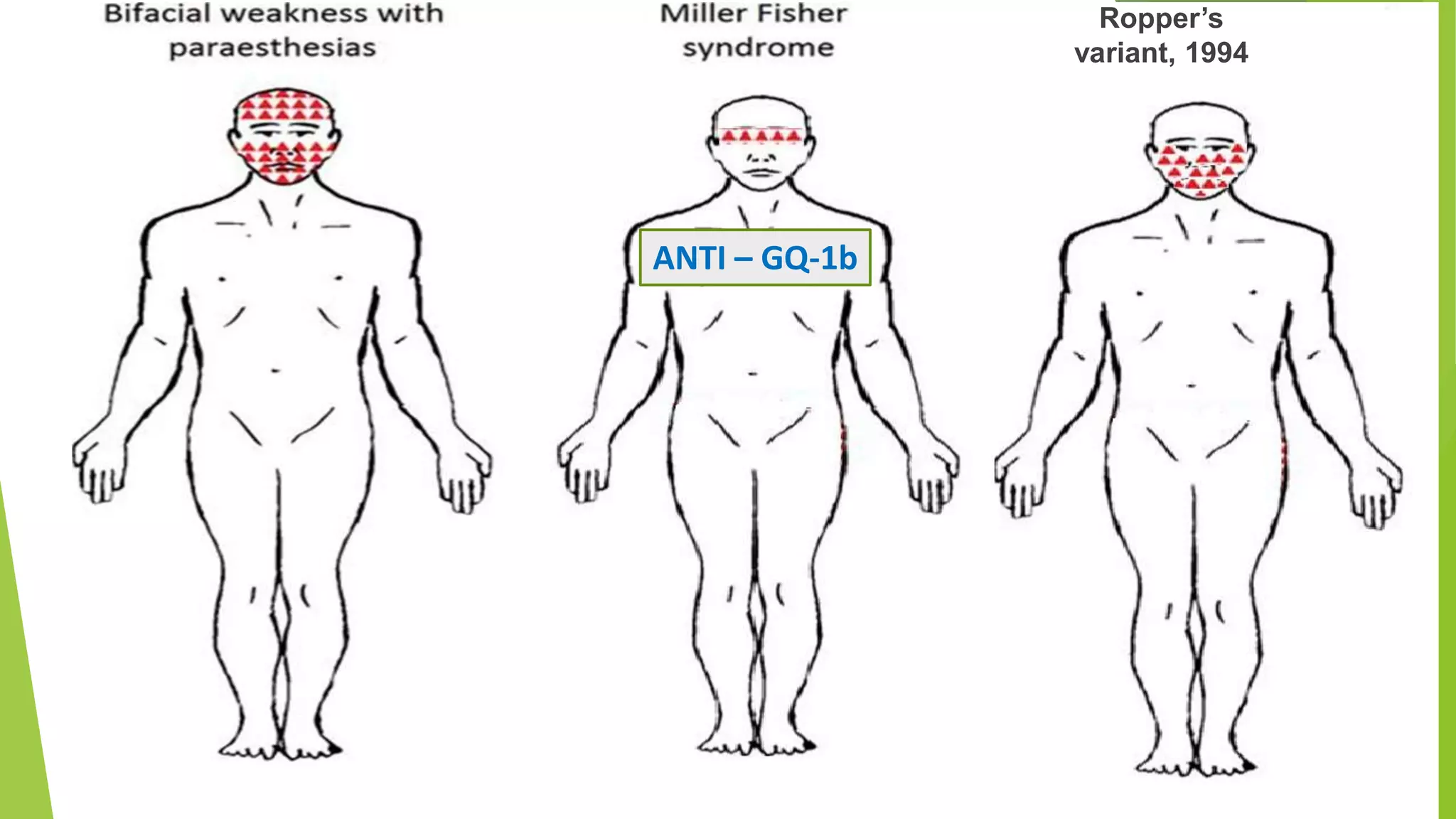





















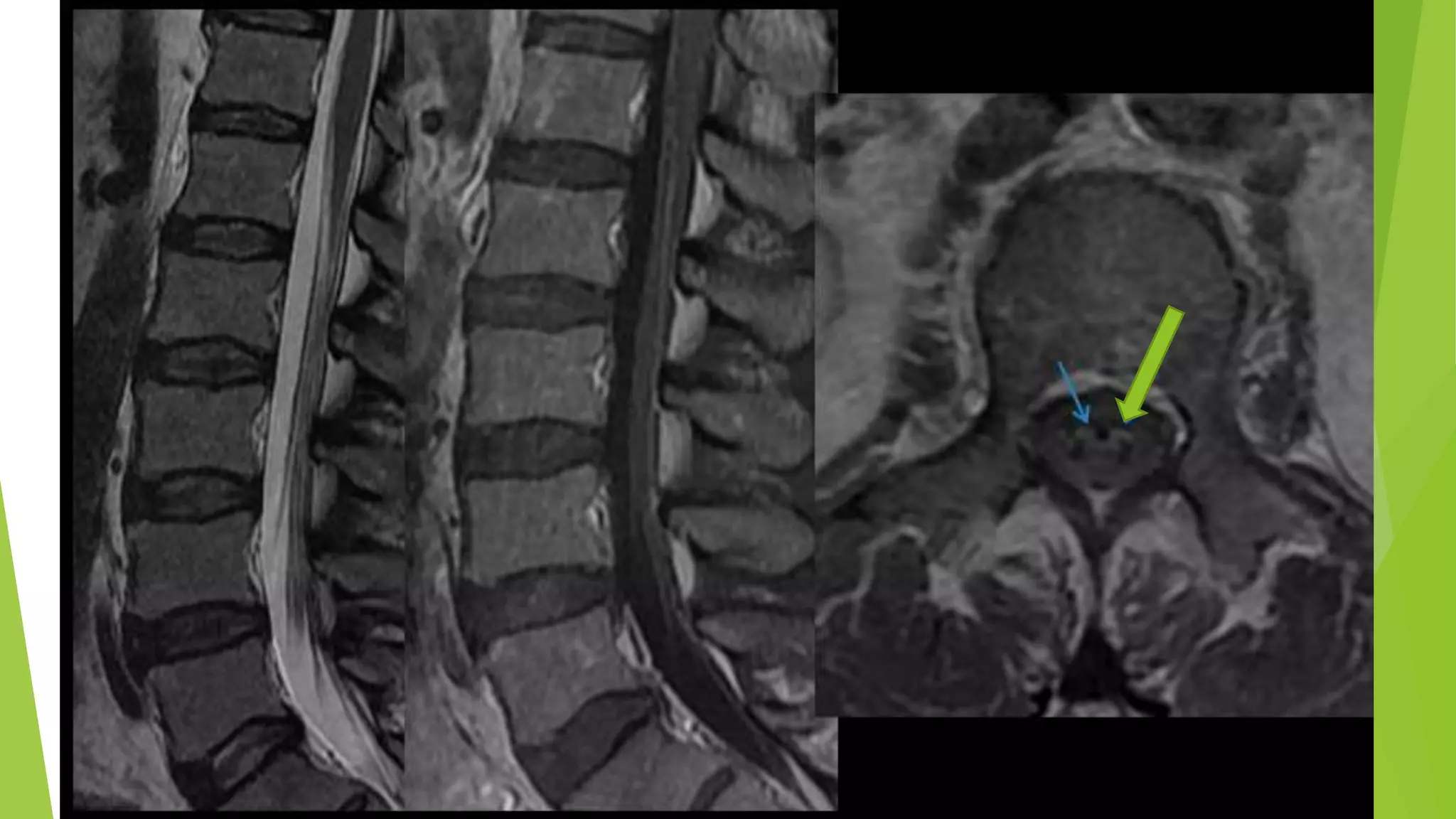


















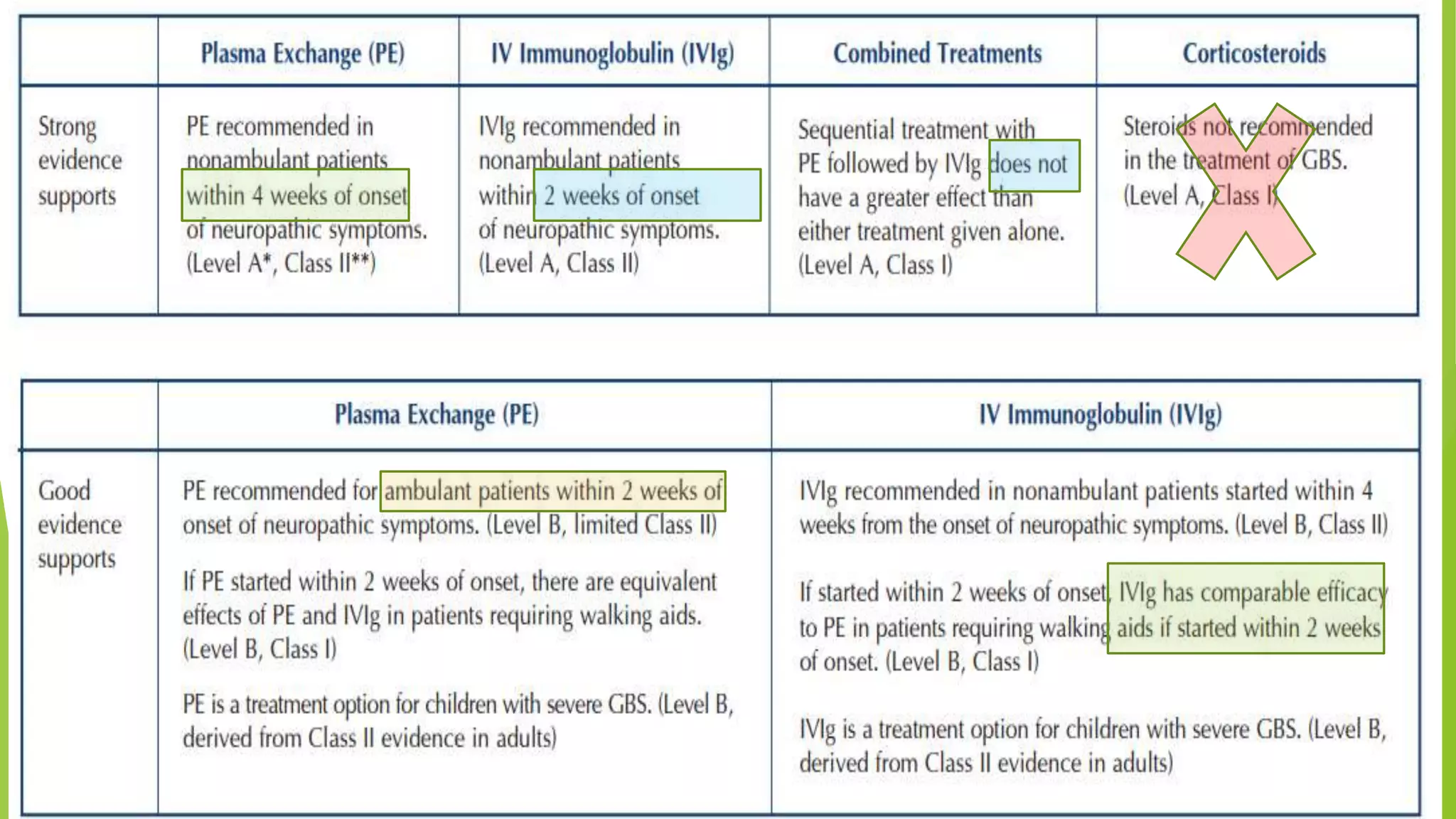












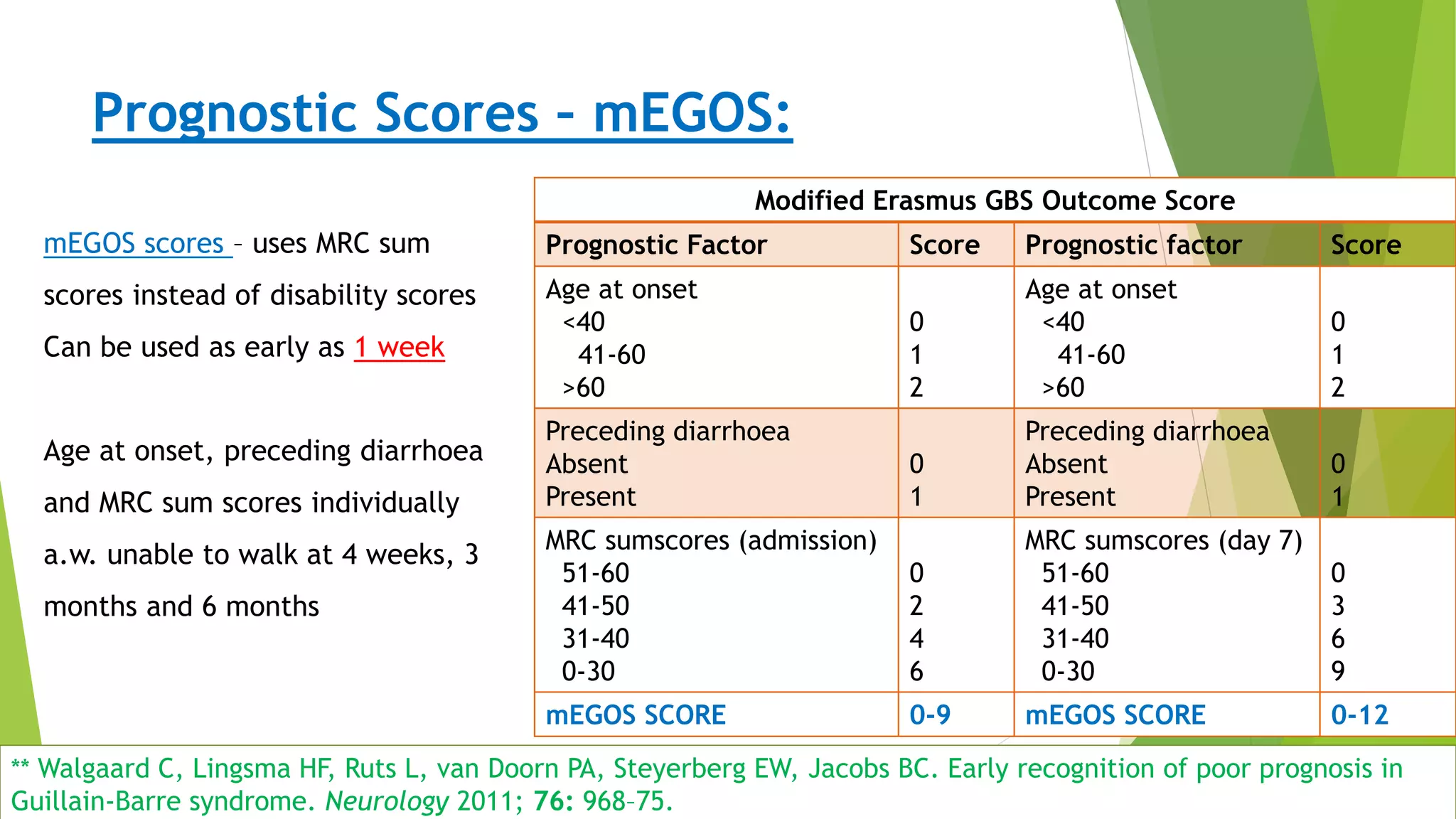



1. Immune-mediated polyneuropathy refers to conditions like Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyneuropathy where the peripheral nerves are damaged by the immune system. 2. GBS is the most common acute monophasic polyneuropathy and is characterized by acute onset of ascending areflexic weakness due to demyelination of peripheral nerves. 3. Diagnosis of GBS involves lumbar puncture showing elevated cerebrospinal fluid protein levels without an increase in white blood cells, along with clinical features of progressive ascending weakness.














































































