Download to read offline

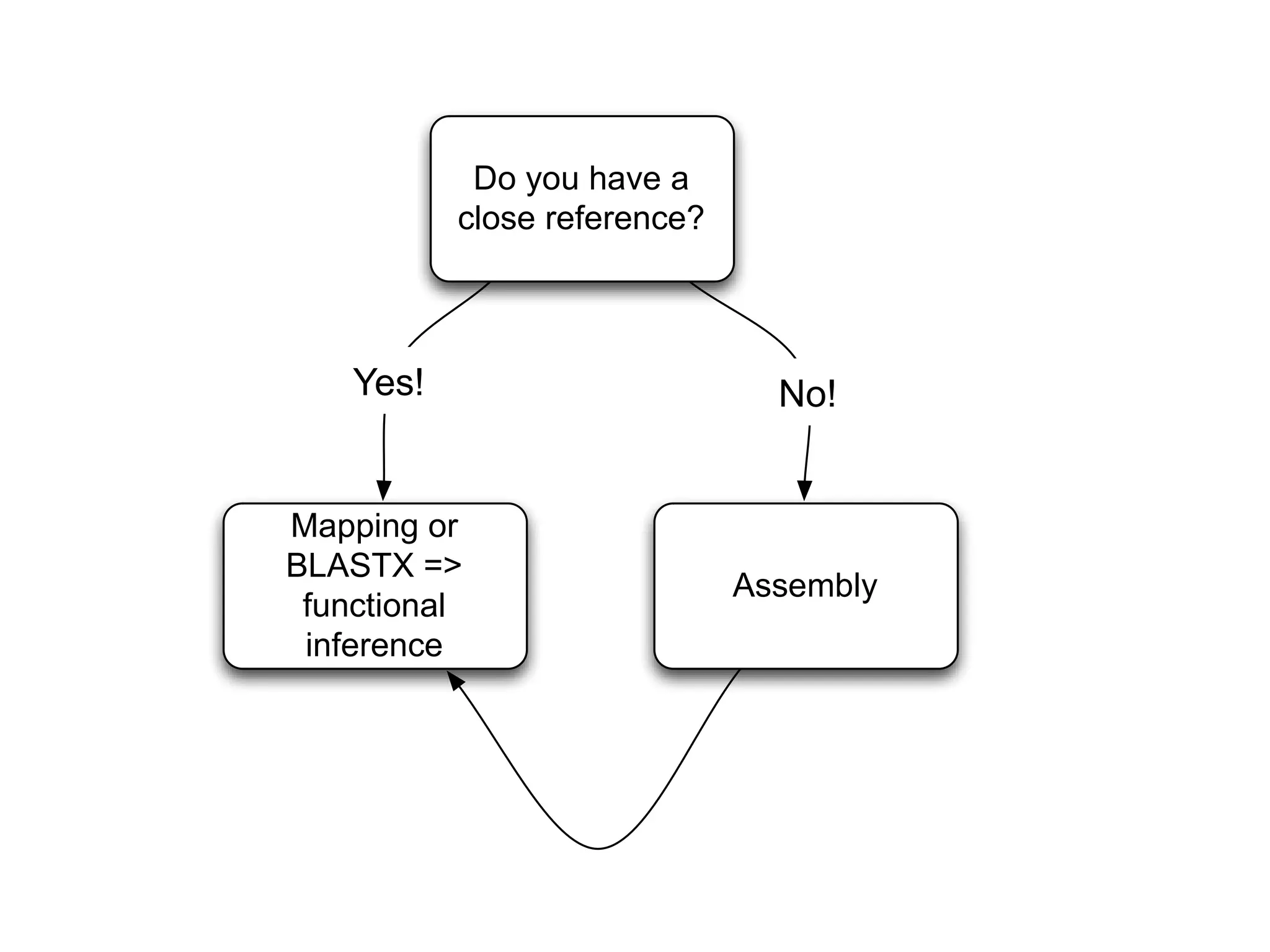



This document outlines the steps in the Kalamazoo Metagenomics Pipeline for analyzing metagenomic sequencing data. The steps include: 1) adapter trimming and quality filtering of reads, 2) normalization of read coverage using Diginorm, 3) partitioning reads into groups and re-inflating groups, 4) assembling reads into contigs, 5) mapping reads back to the assembly, and 6) annotating contigs with abundance information using tools like MG-RAST.


































































