Downloaded 115 times



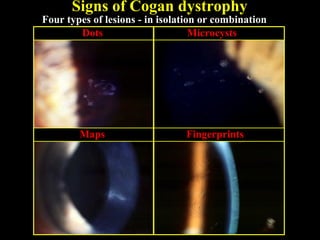
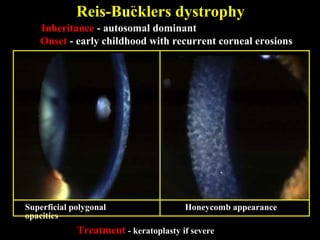
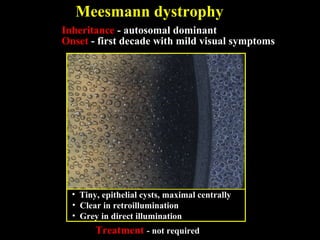
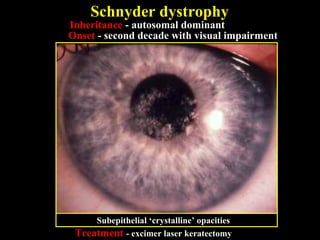
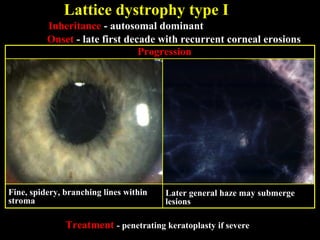
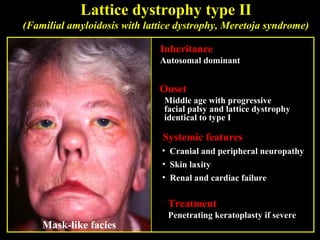

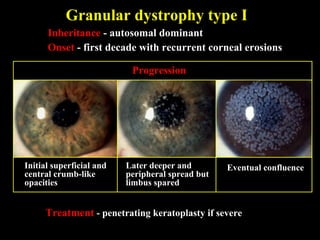

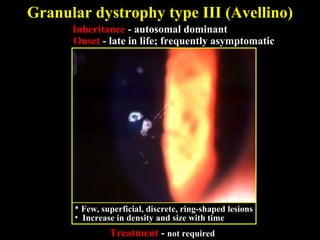
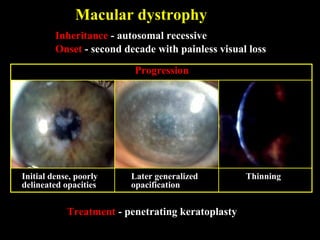
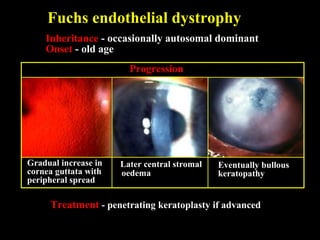
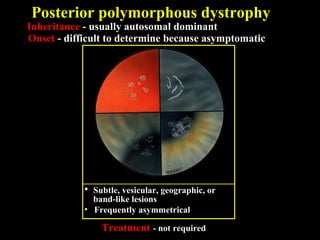

This document summarizes various types of corneal dystrophies: - Anterior corneal dystrophies include Cogan microcystic, Reis-Bucklers, Meesmann, and Schnyder dystrophies. - Stromal dystrophies include lattice types I-III and granular types I-III (Avellino). - Posterior dystrophies include Fuchs endothelial and posterior polymorphous dystrophies. Each dystrophy is described in terms of inheritance, onset, symptoms, signs, treatment or progression.









































































































