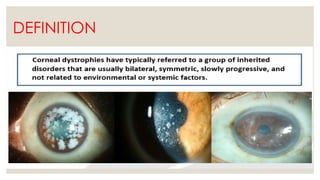
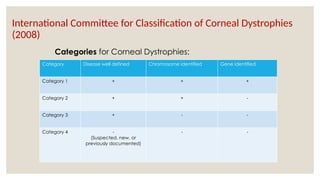
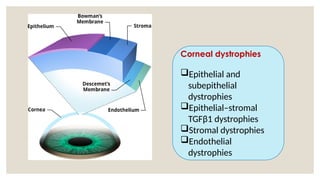
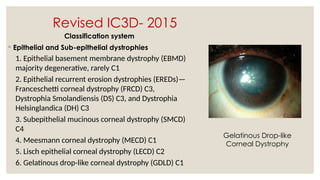
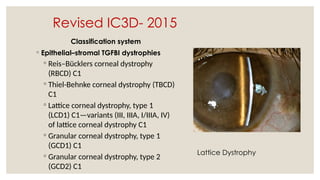
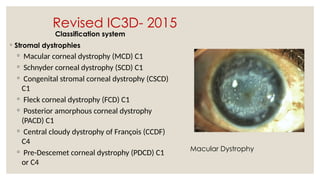
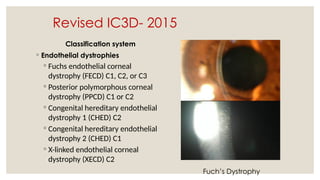
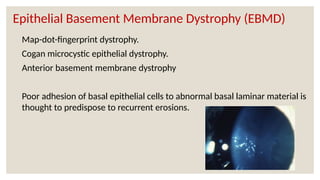
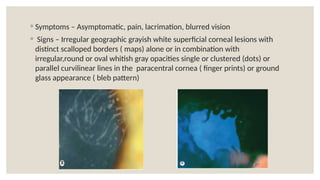
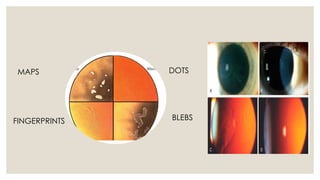
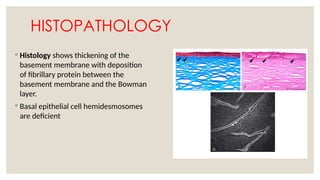
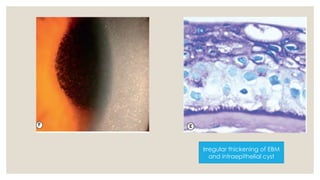
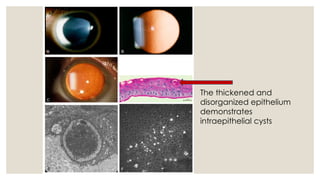
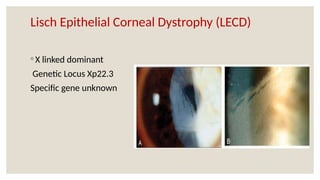
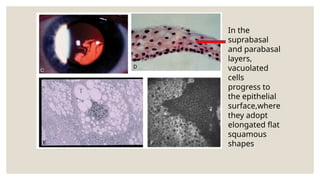
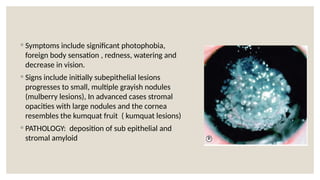
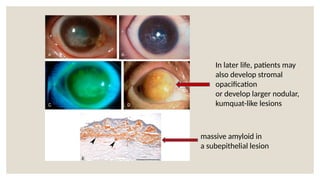
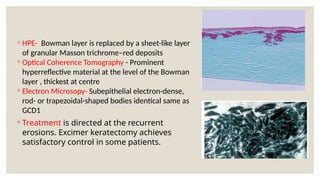
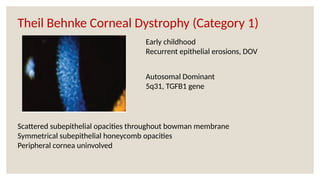
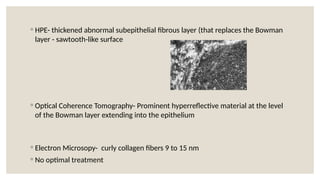
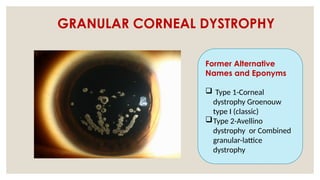
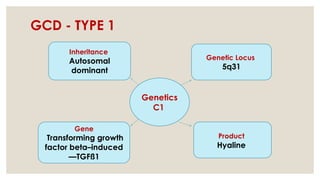
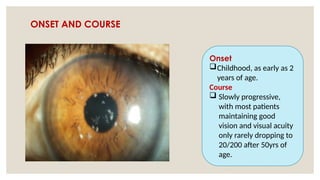
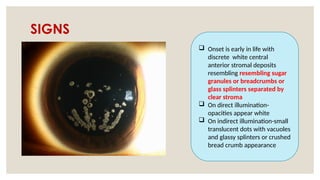
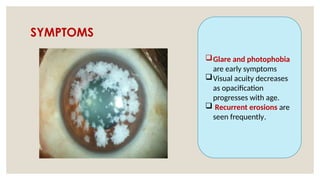
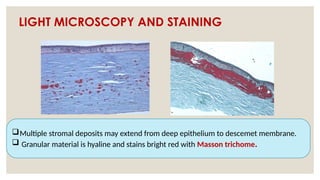
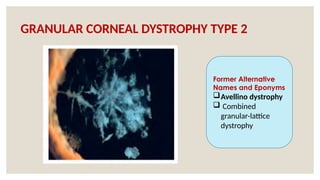
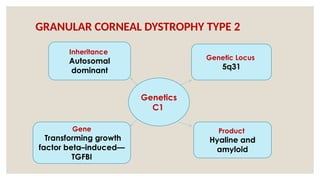
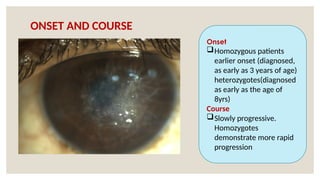
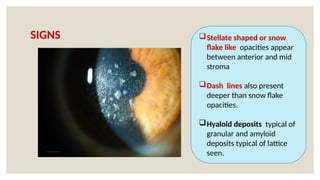
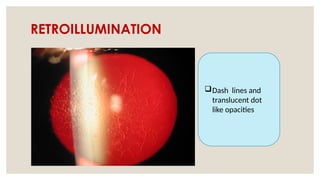
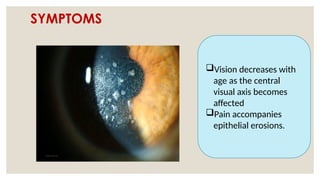
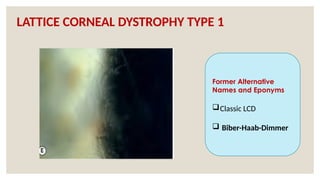
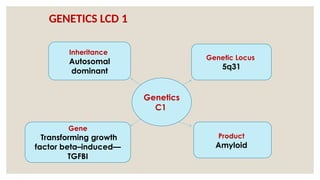
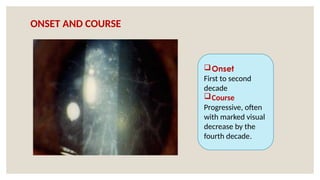
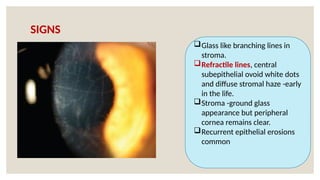
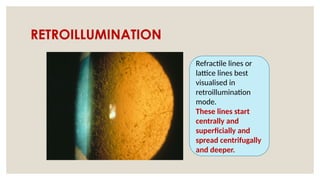
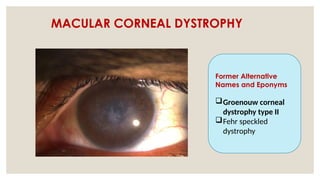
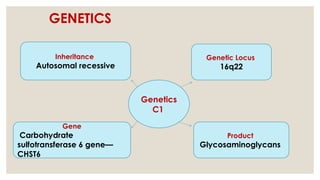
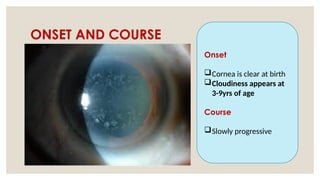
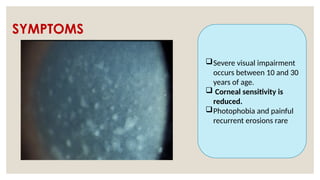
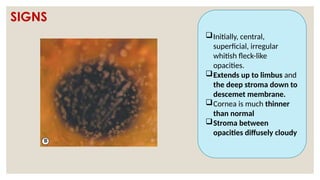
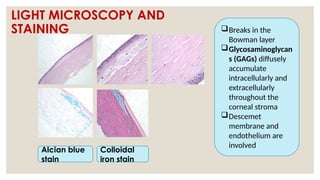
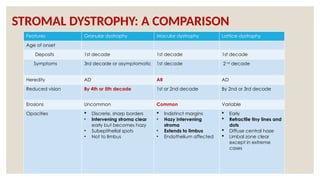
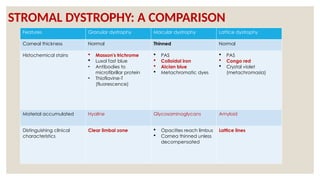
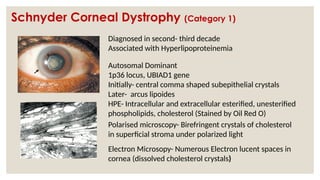
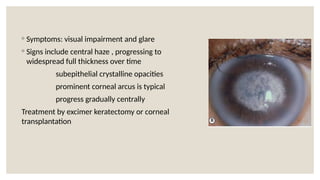
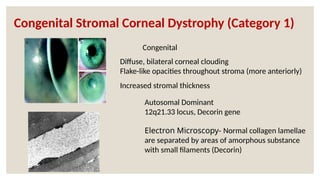
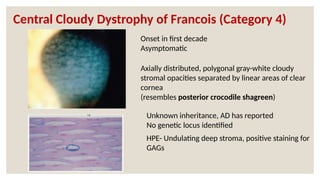
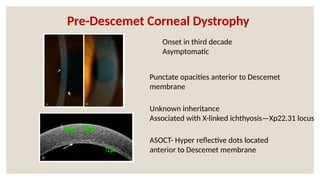
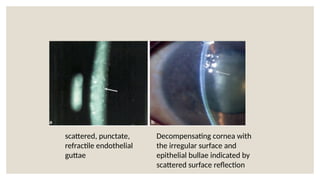
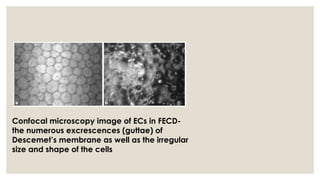
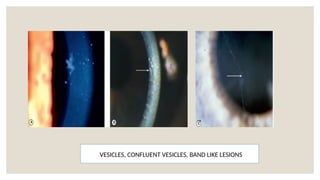
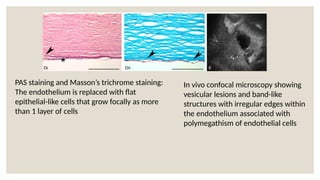
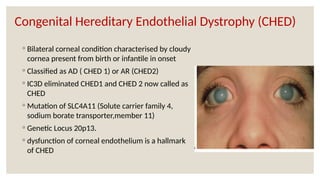
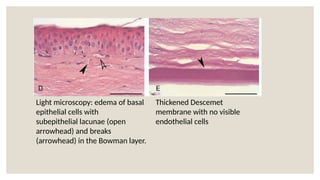
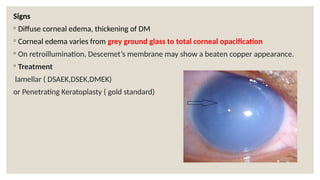
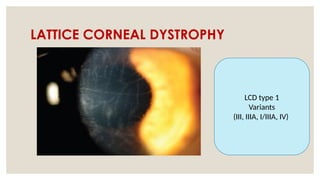
The document provides a detailed overview of corneal dystrophies, including their classifications, genetic factors, and clinical features. It highlights the evolution of the International Committee for Classification of Corneal Dystrophies (IC3D) from 2008 to 2015, focusing on anatomical and histopathological considerations. The summary includes specific types of dystrophies, their symptoms, inheritance patterns, and management options.