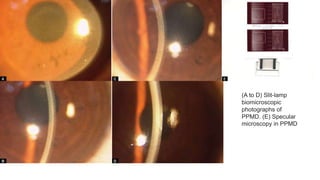
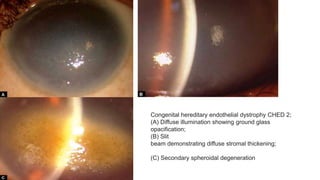
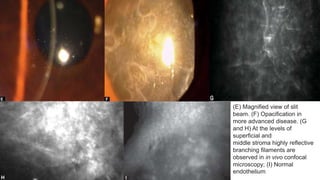
This document provides information on several types of corneal dystrophies:
- Epithelial basement membrane dystrophy is usually asymptomatic and presents in adulthood with recurrent erosion. MAPS, DOTS, and fingerprint lines are seen on examination. Treatments include debridement and bandage contact lenses.
- Epithelial recurrent erosion dystrophy presents in childhood with attacks of pain and photophobia. Corneal erosions are seen during attacks which decline by age 50. Treatments include antibiotics and corticosteroids.
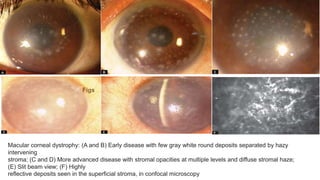
- Stromal dystrophies include lattice, granular, and macular corneal dystrophies which are progressive and cause visual impairment. Signs include characteristic opacities seen on slit lamp









![• MEESMAN CORNEAL DYSTROPHY
• Inheritance: Autosomal dominant
• Onset,course,symptom:
• • occurs in early childhood & is slowly progressive with variant stocker holt dystrophy with
• mild visual reduction
• • Patient complains glare & light sensitivity, recurrent painful epithelial erosions.
• • Rarely blurred vision results from corneal irregularity & scarring.
• • PATHOLOGY: It has been associated with genes KRT3 and KRT12 located on chromosome 12
• and 17 respectively.[4]](https://image.slidesharecdn.com/cornealdystrophy-230614191806-a69c8239/85/CORNEAL-DYSTROPHY-pptx-10-320.jpg)



















![• TREATMENT
• • punctal plugs (both upper and
lower)
• Keretoplasty is required at age of
30-40 years.
• Phototherapeutic
keratectomy (PTK) using
• [Excimer laser] can restore and
preserve useful
• visual function for a significant
period of time in
• patients with anterior corneal
dystrophies.[](https://image.slidesharecdn.com/cornealdystrophy-230614191806-a69c8239/85/CORNEAL-DYSTROPHY-pptx-31-320.jpg)