Download as PDF, PPTX

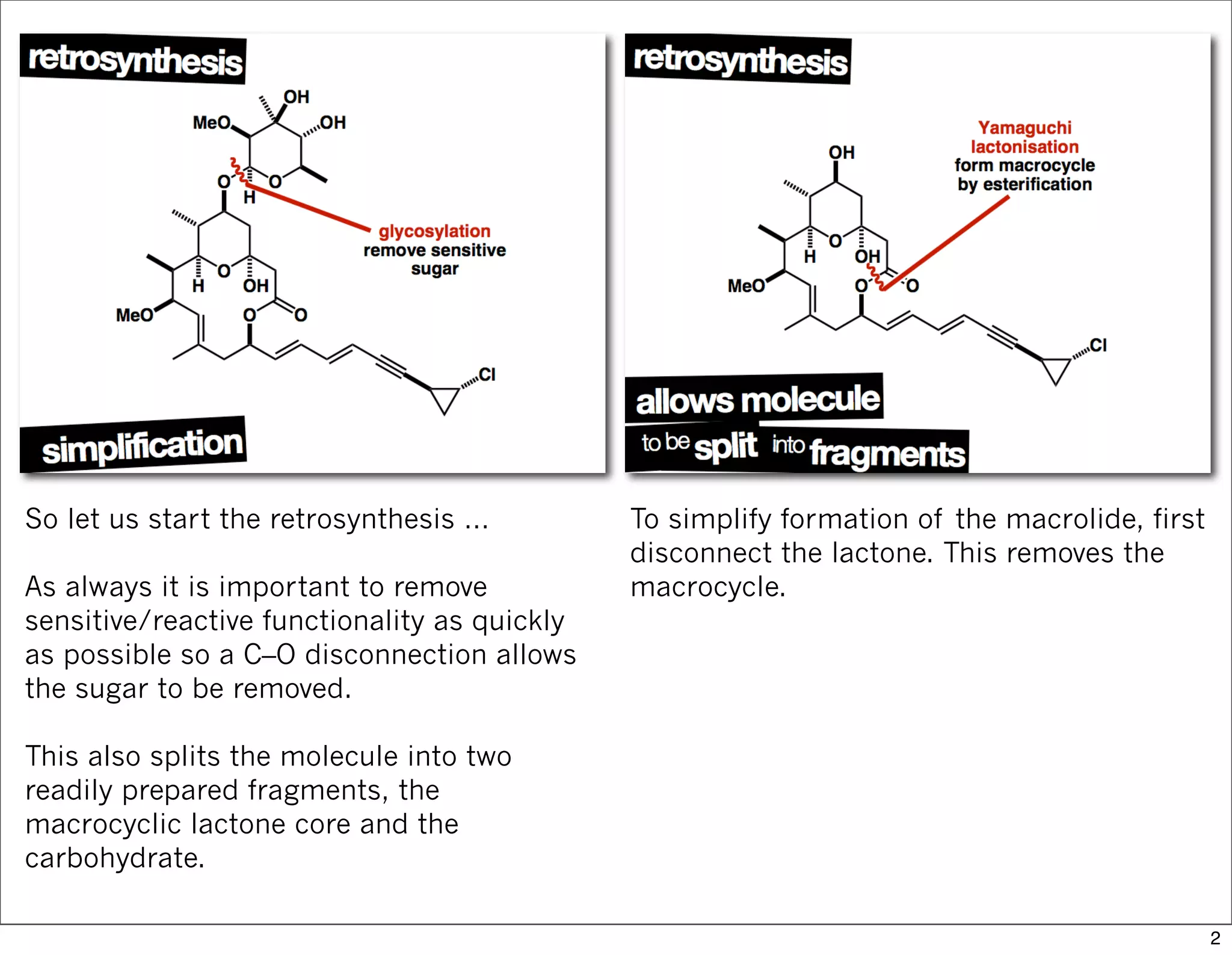
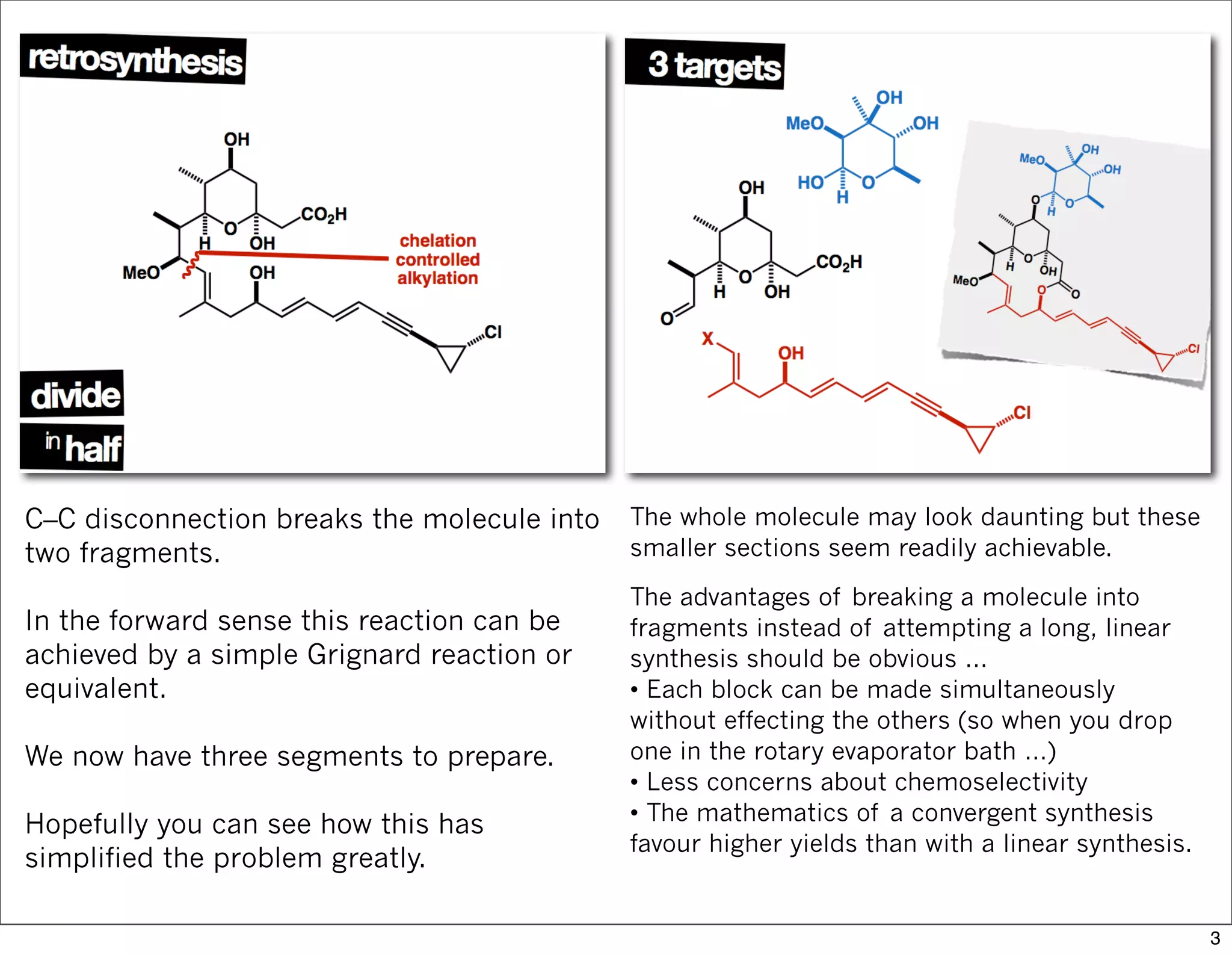
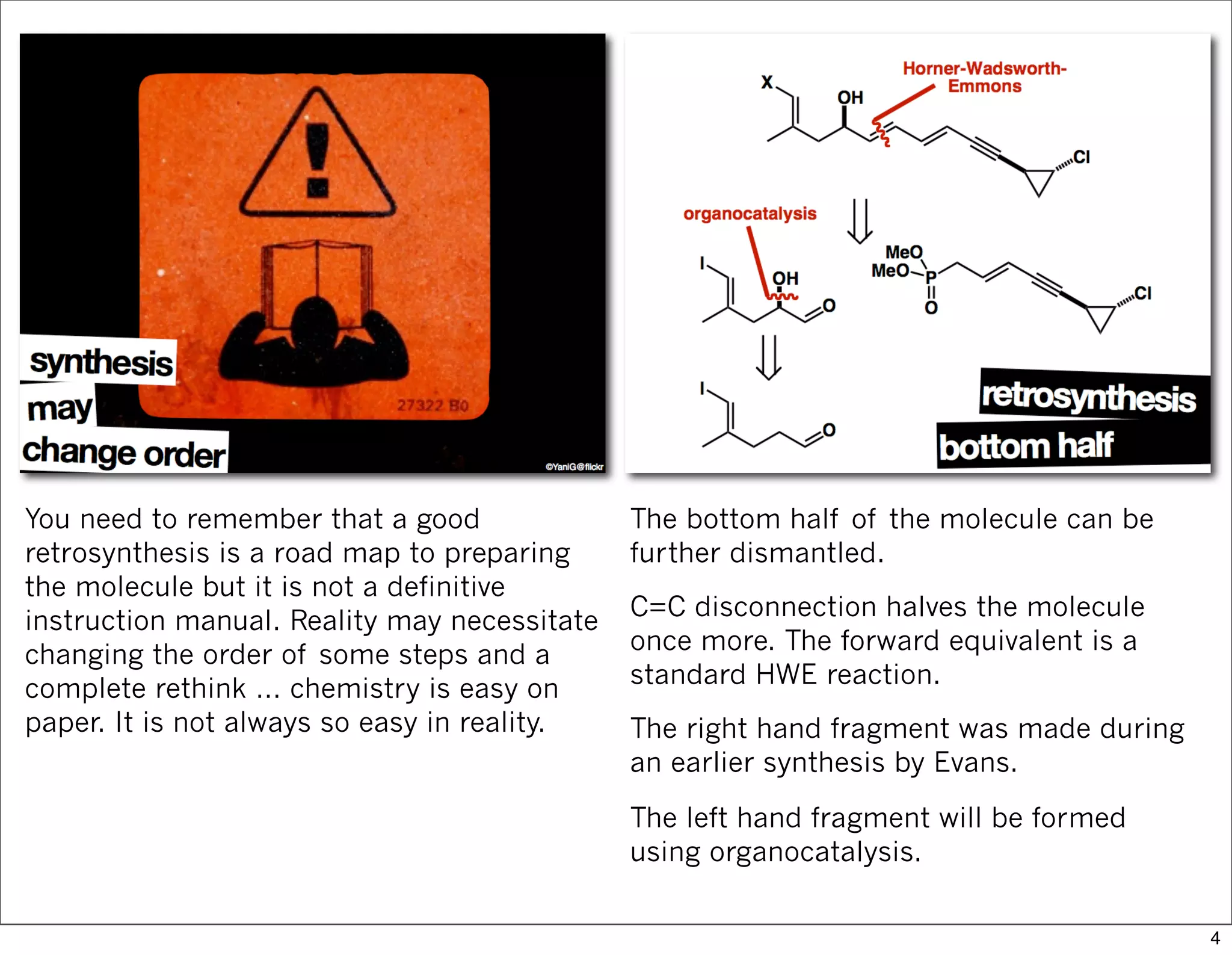
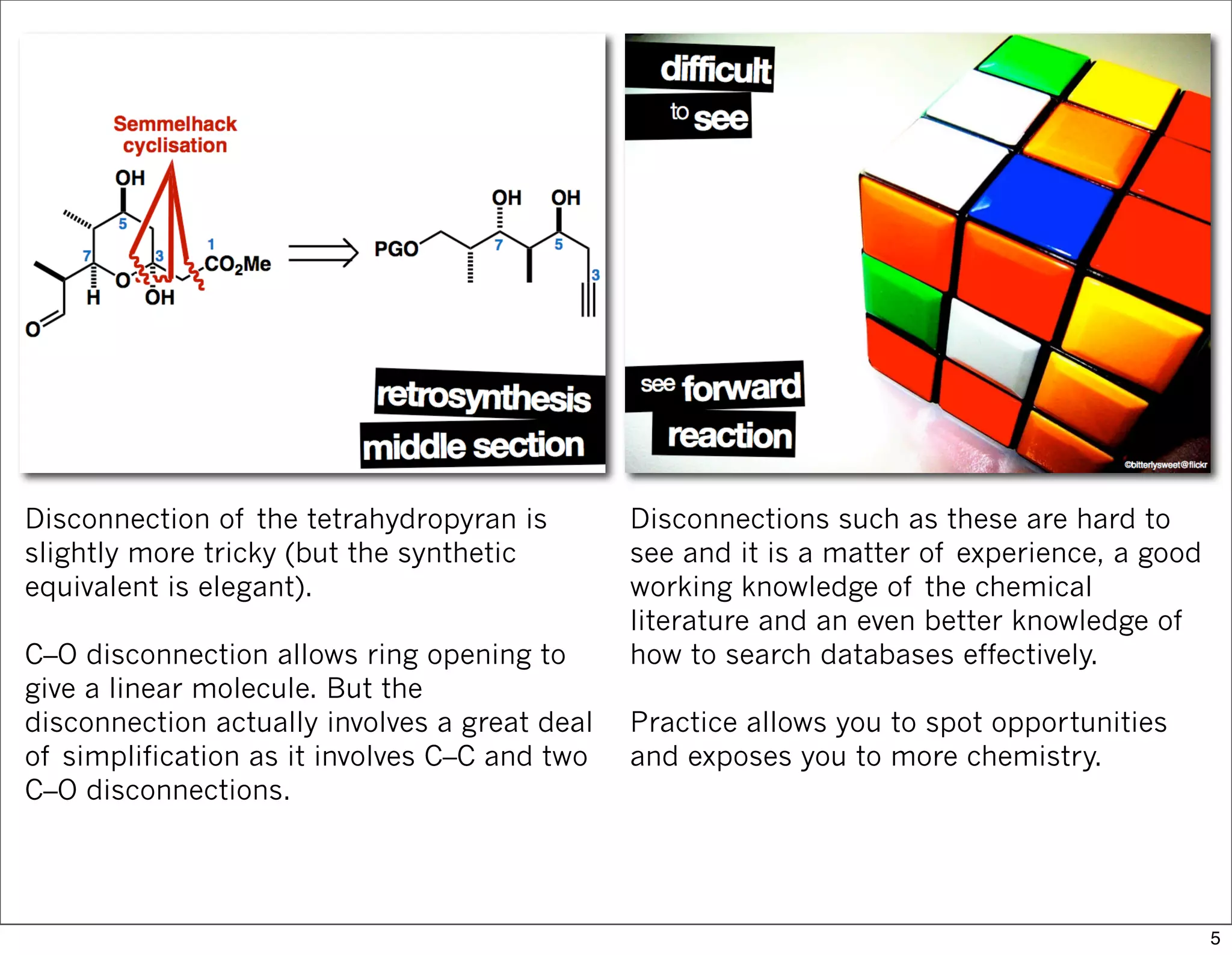
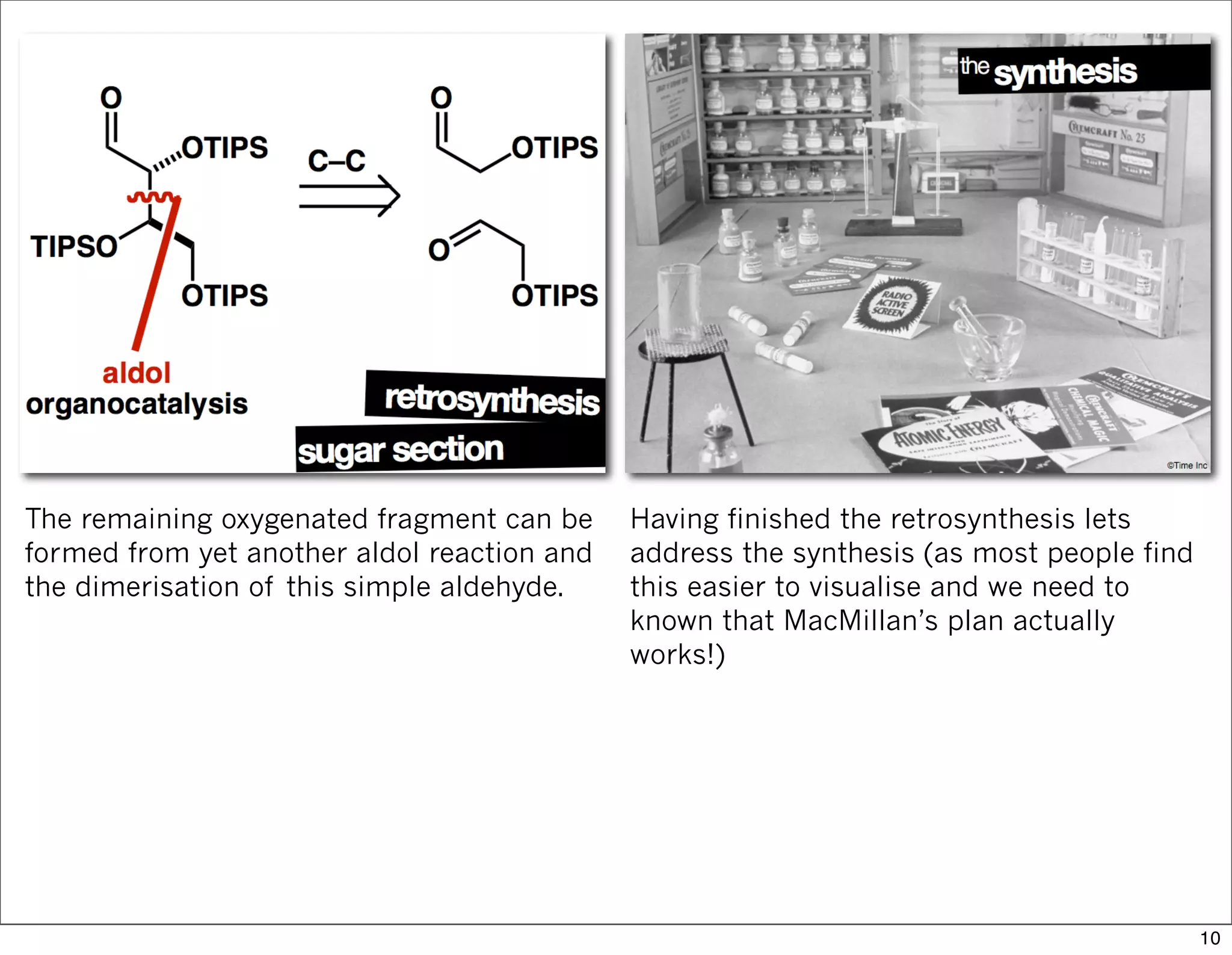
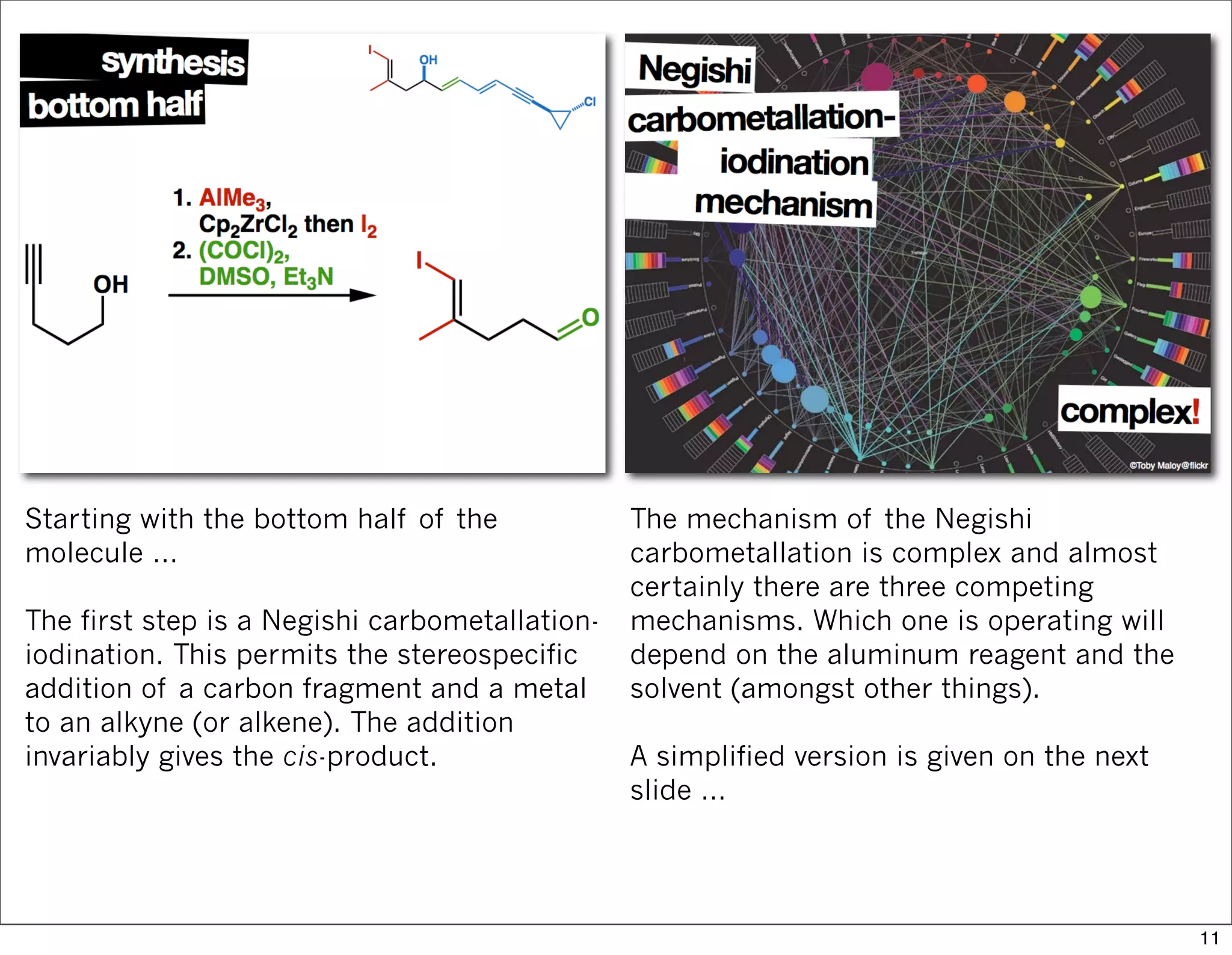
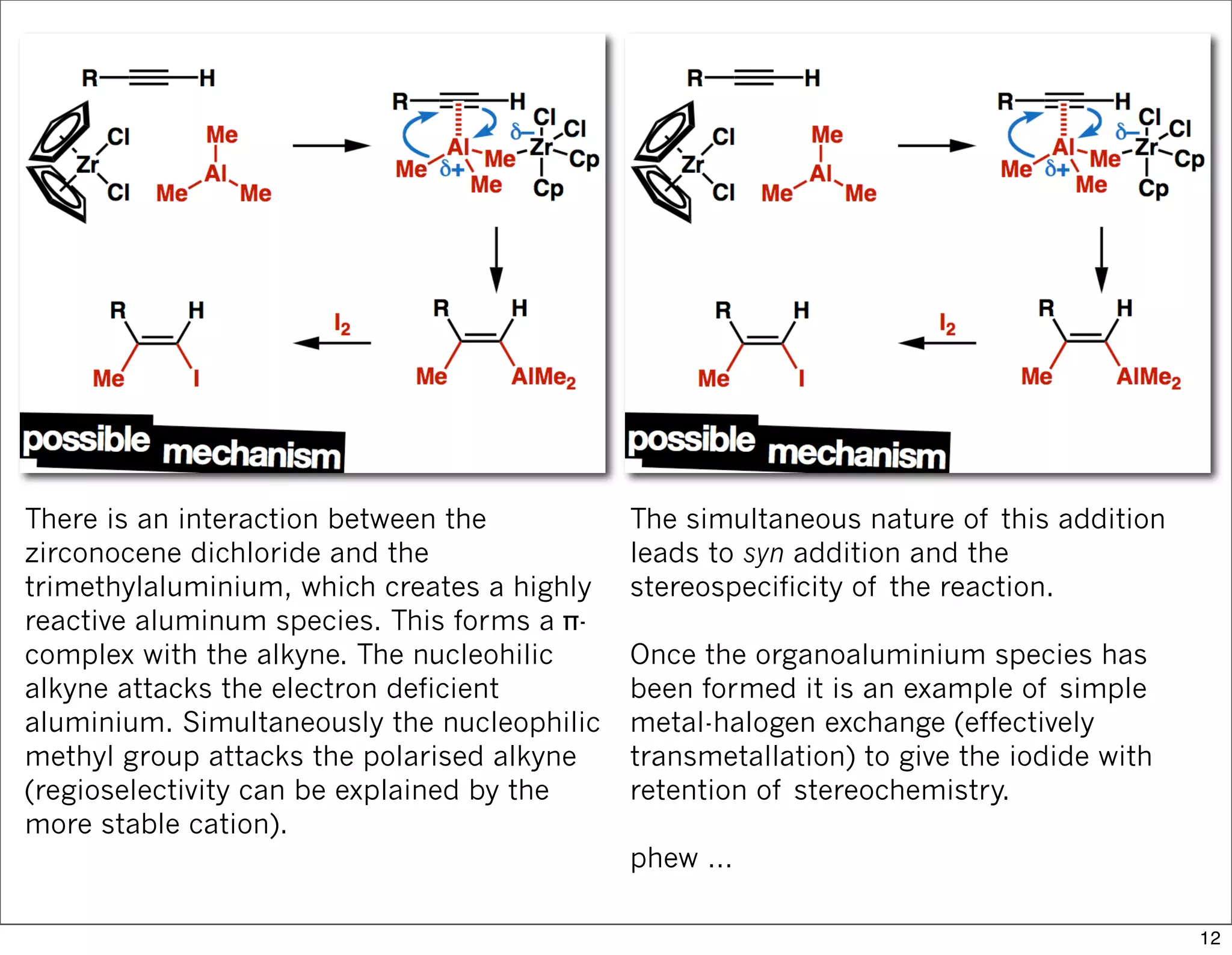
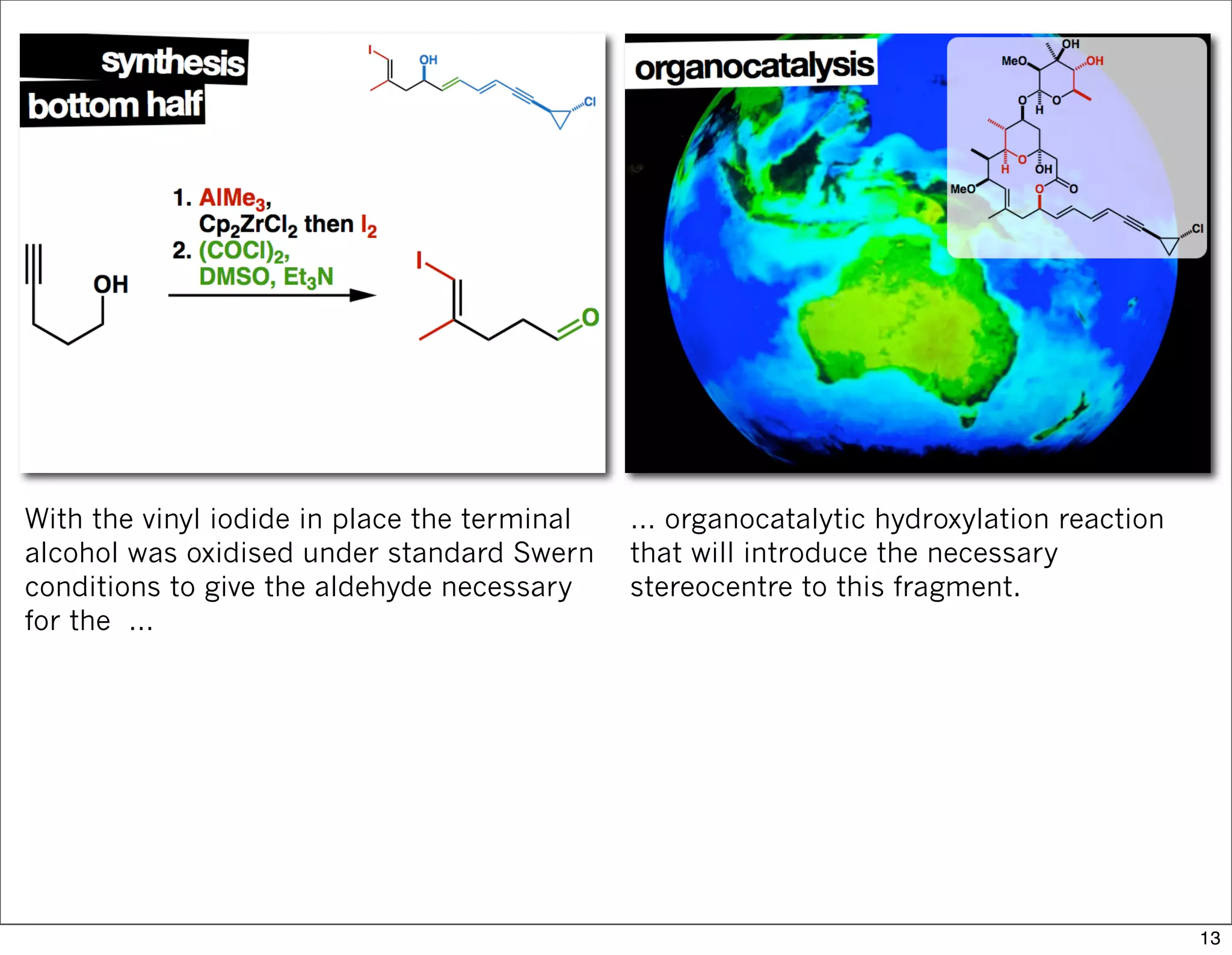
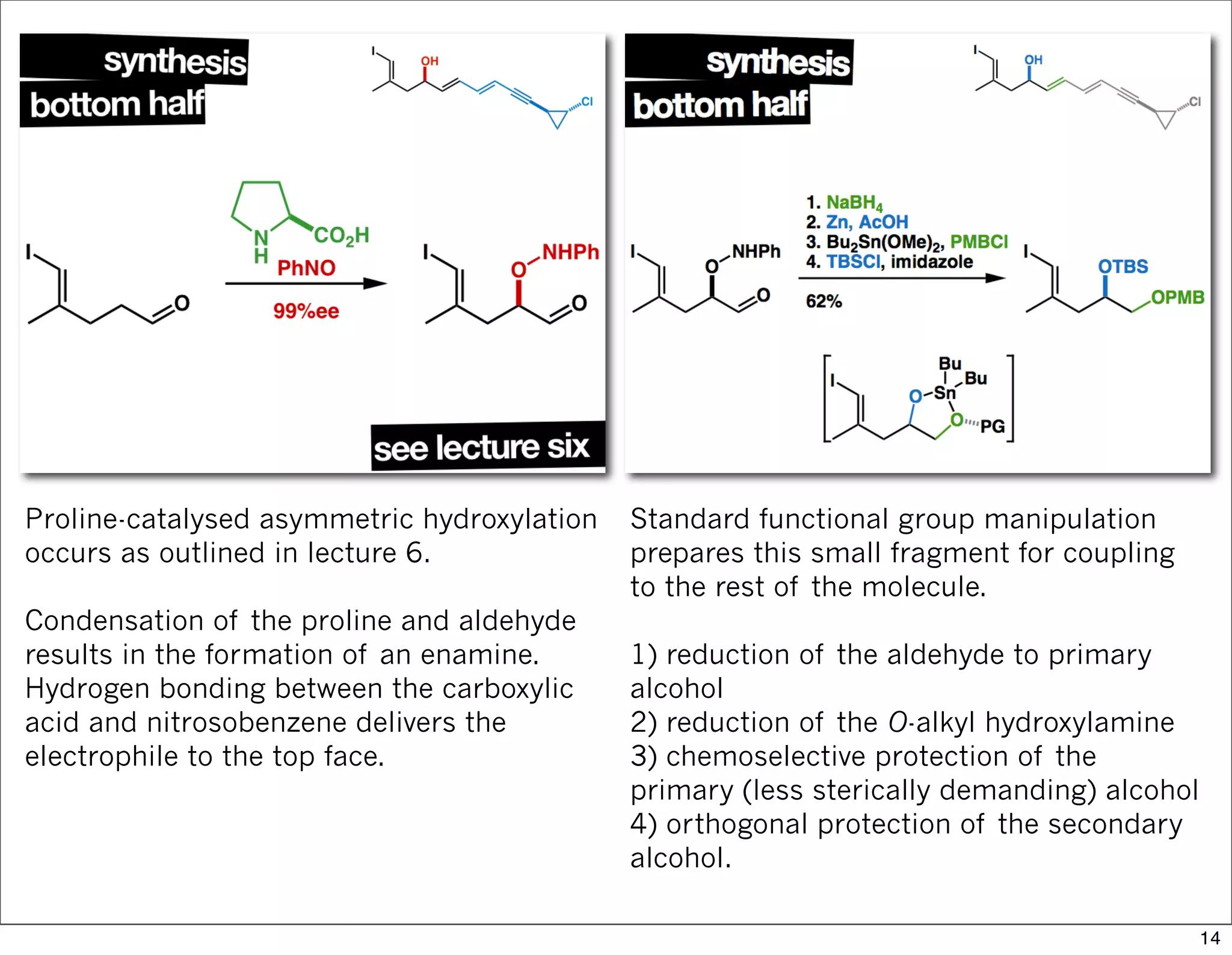
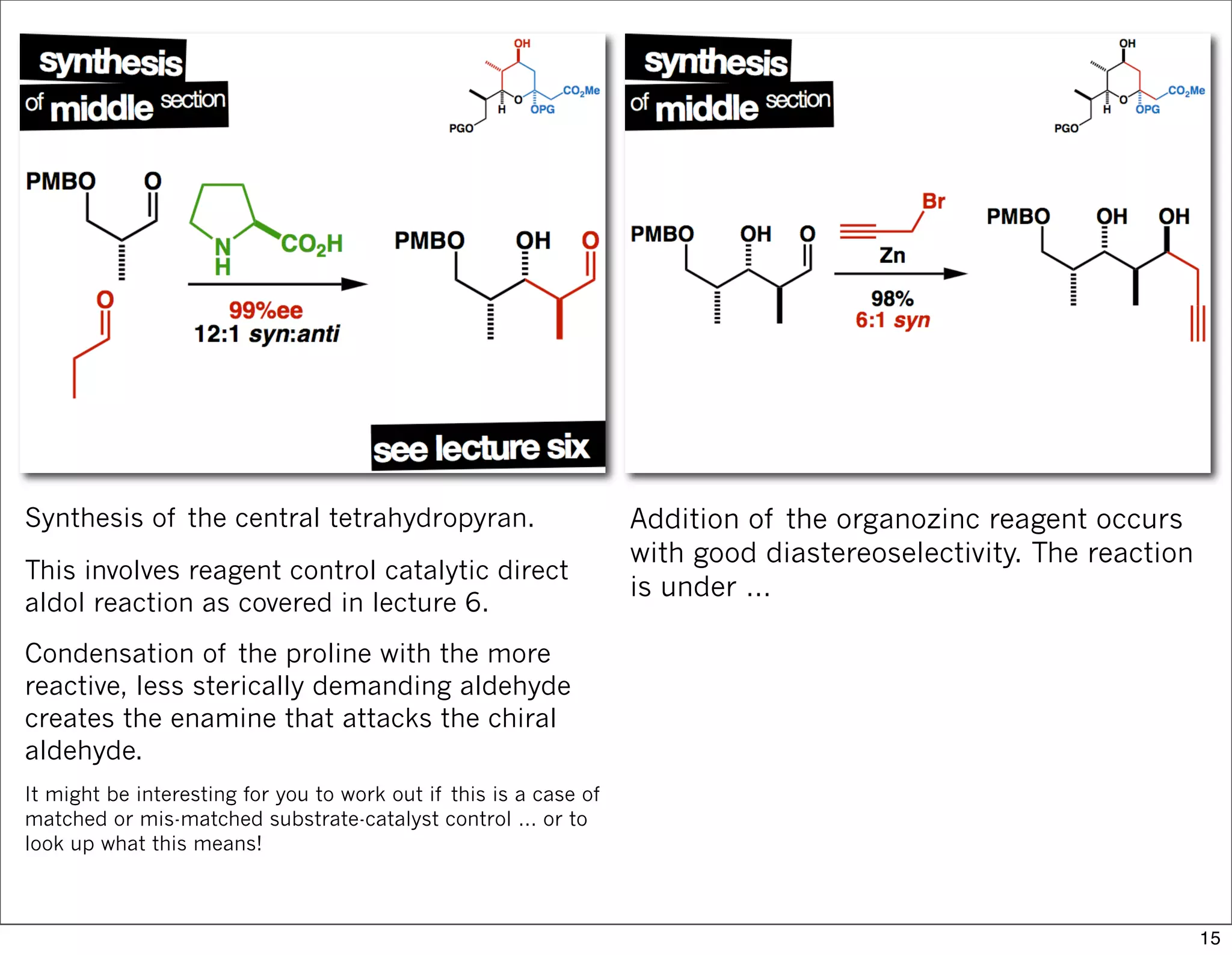
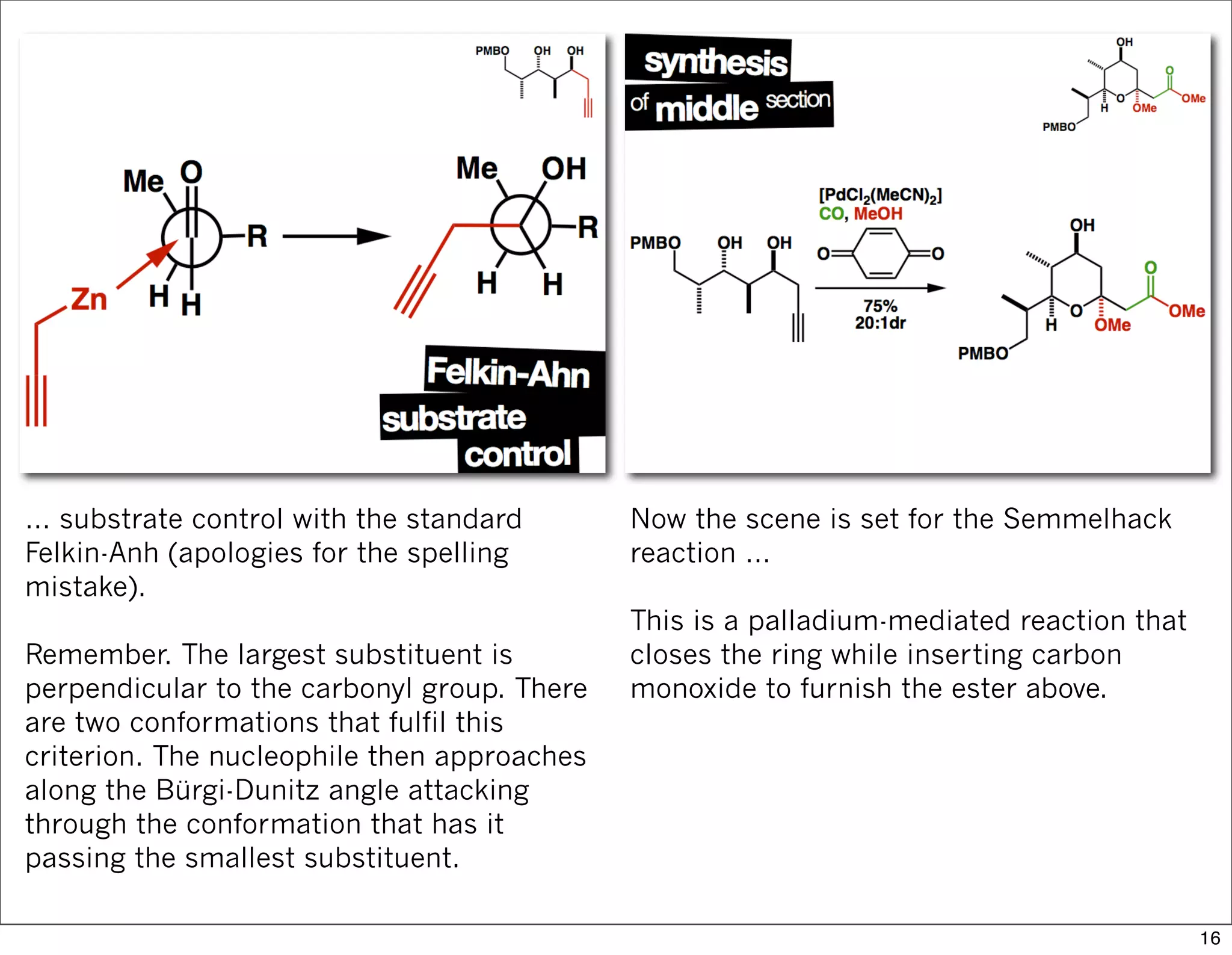

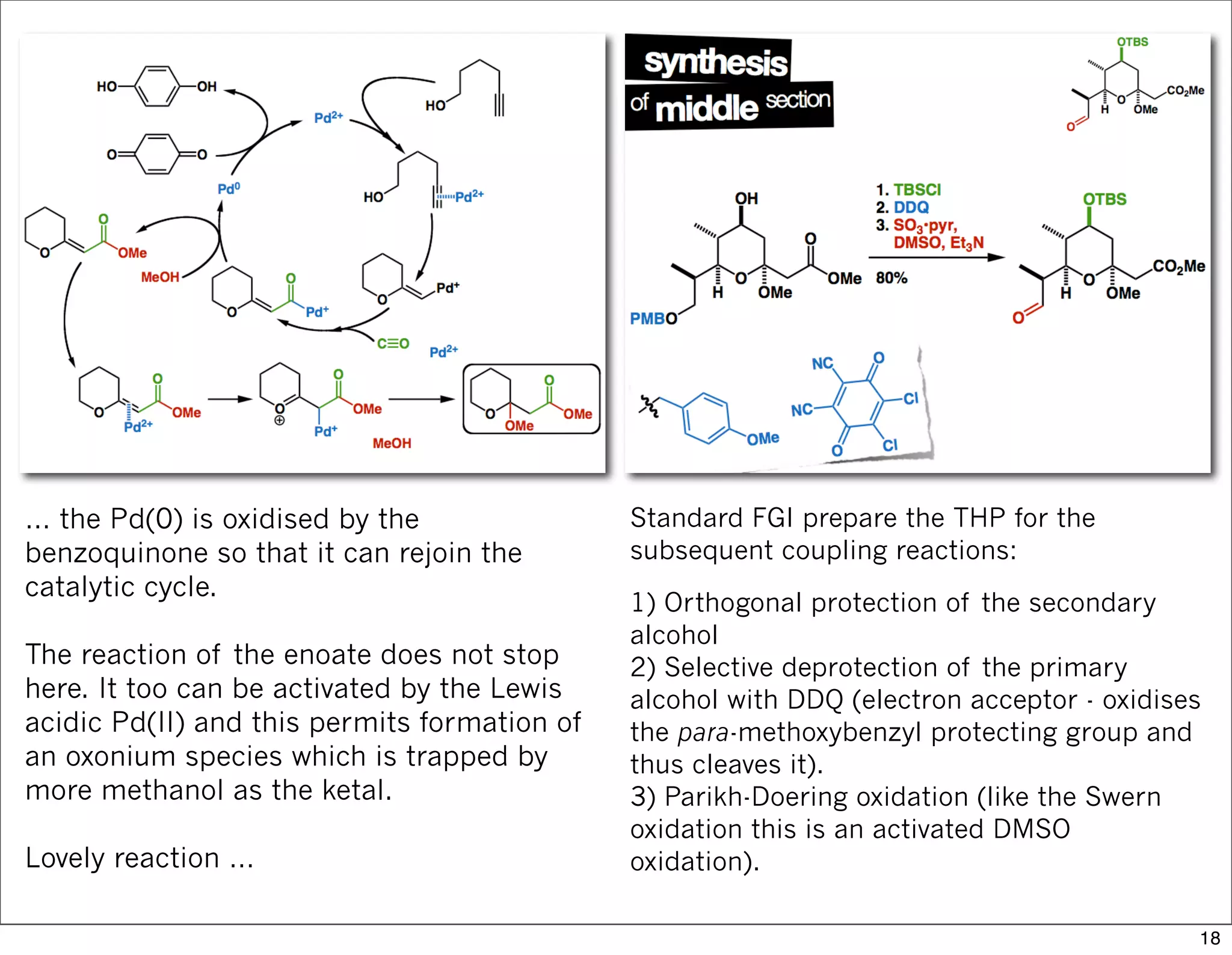
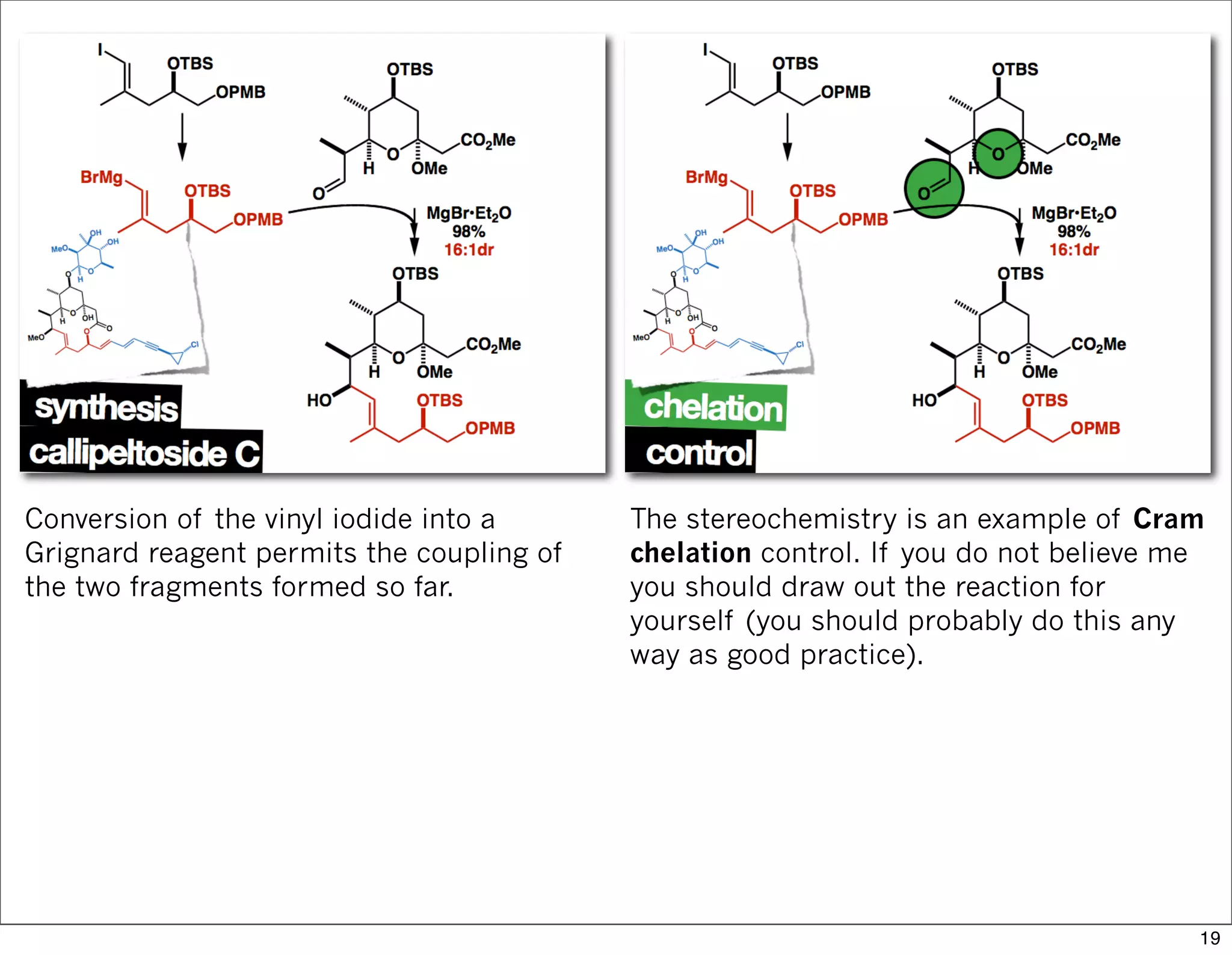
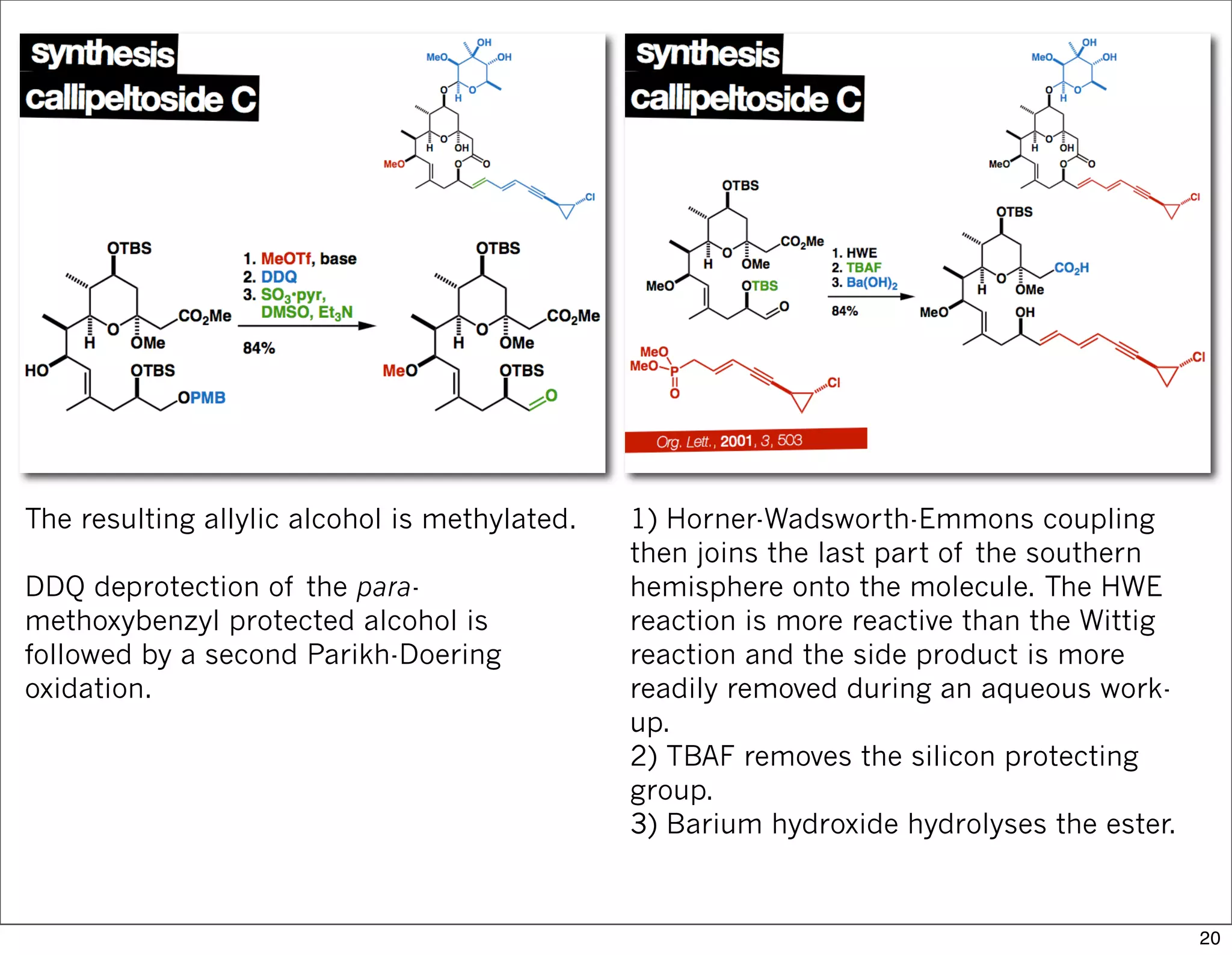
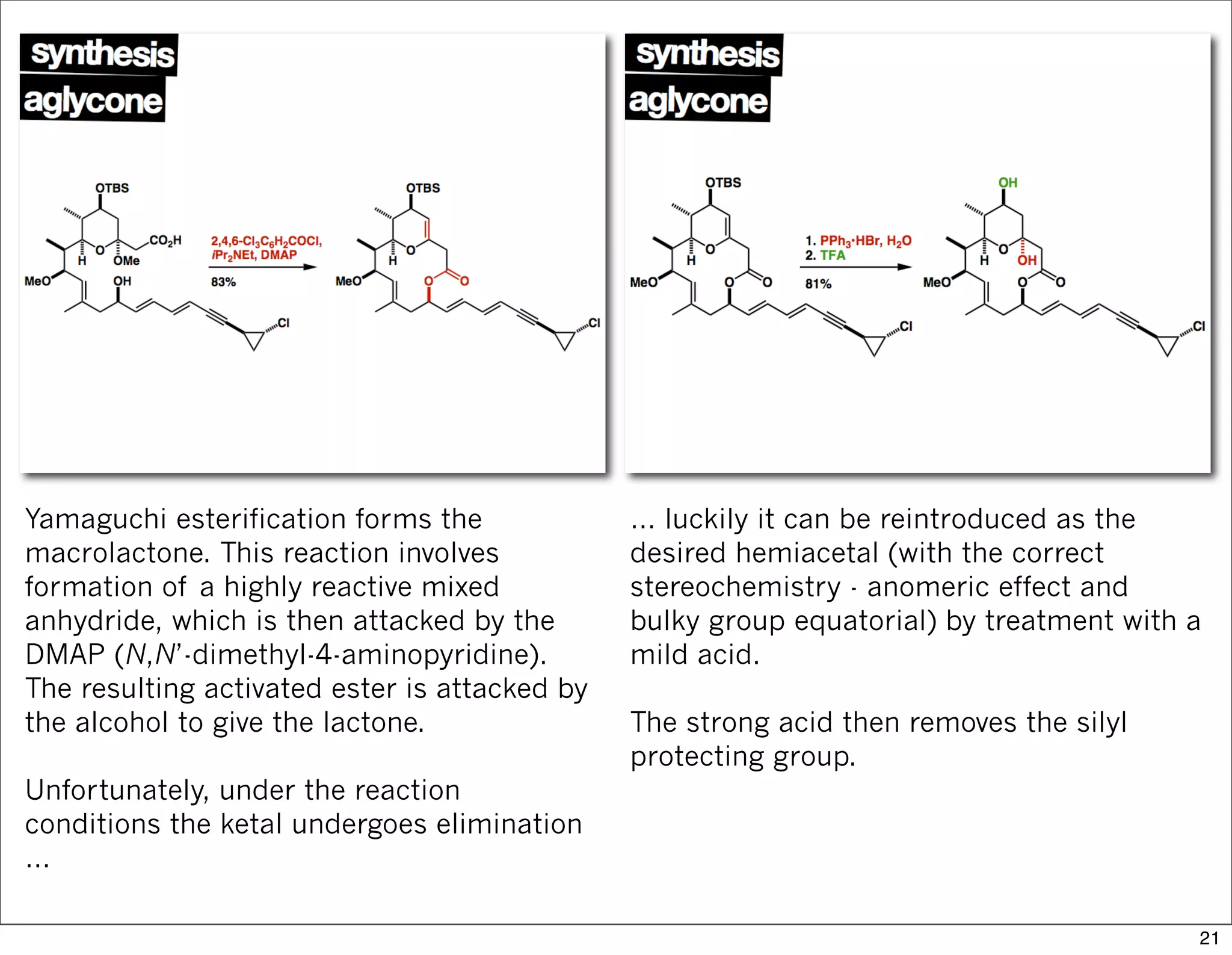

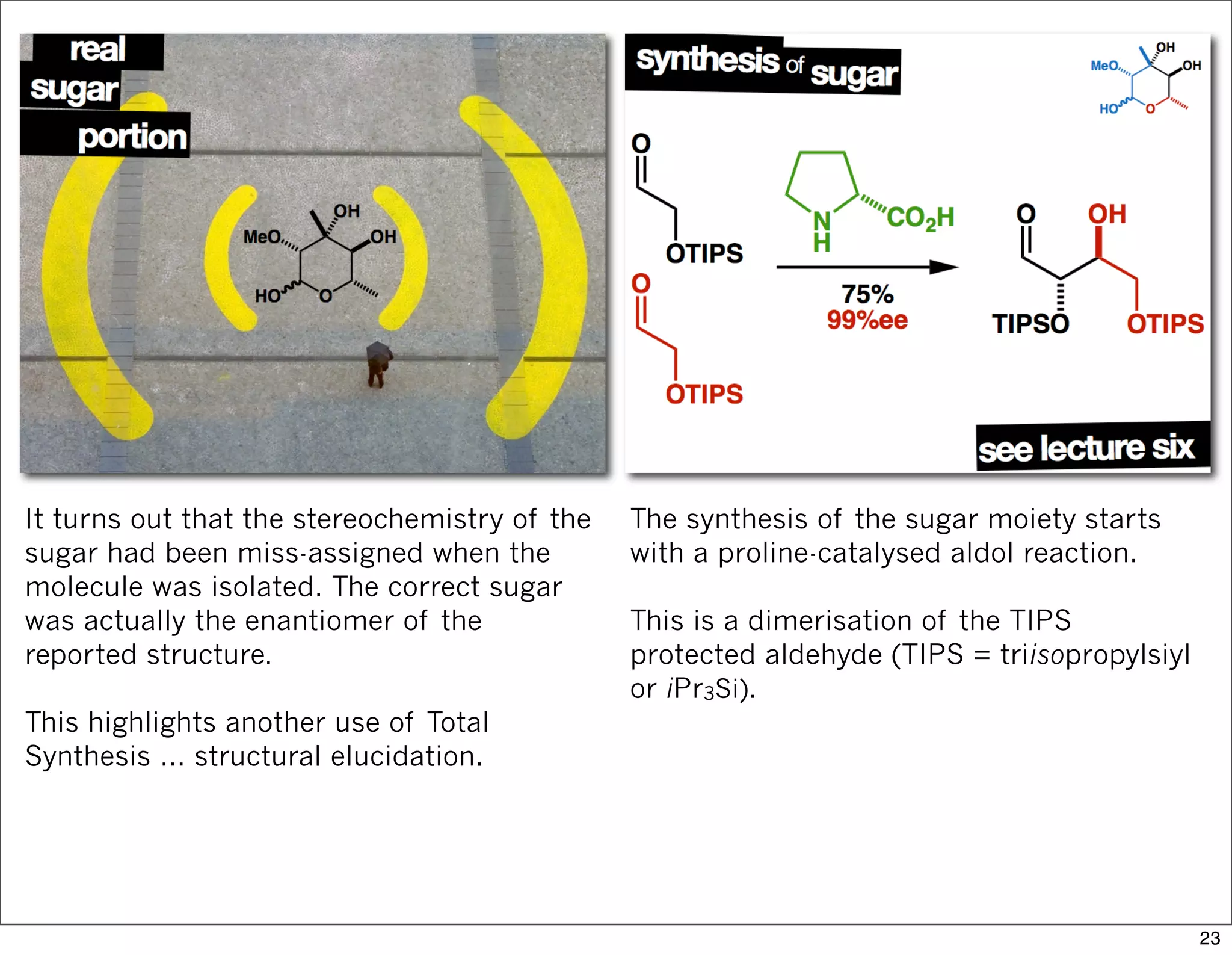
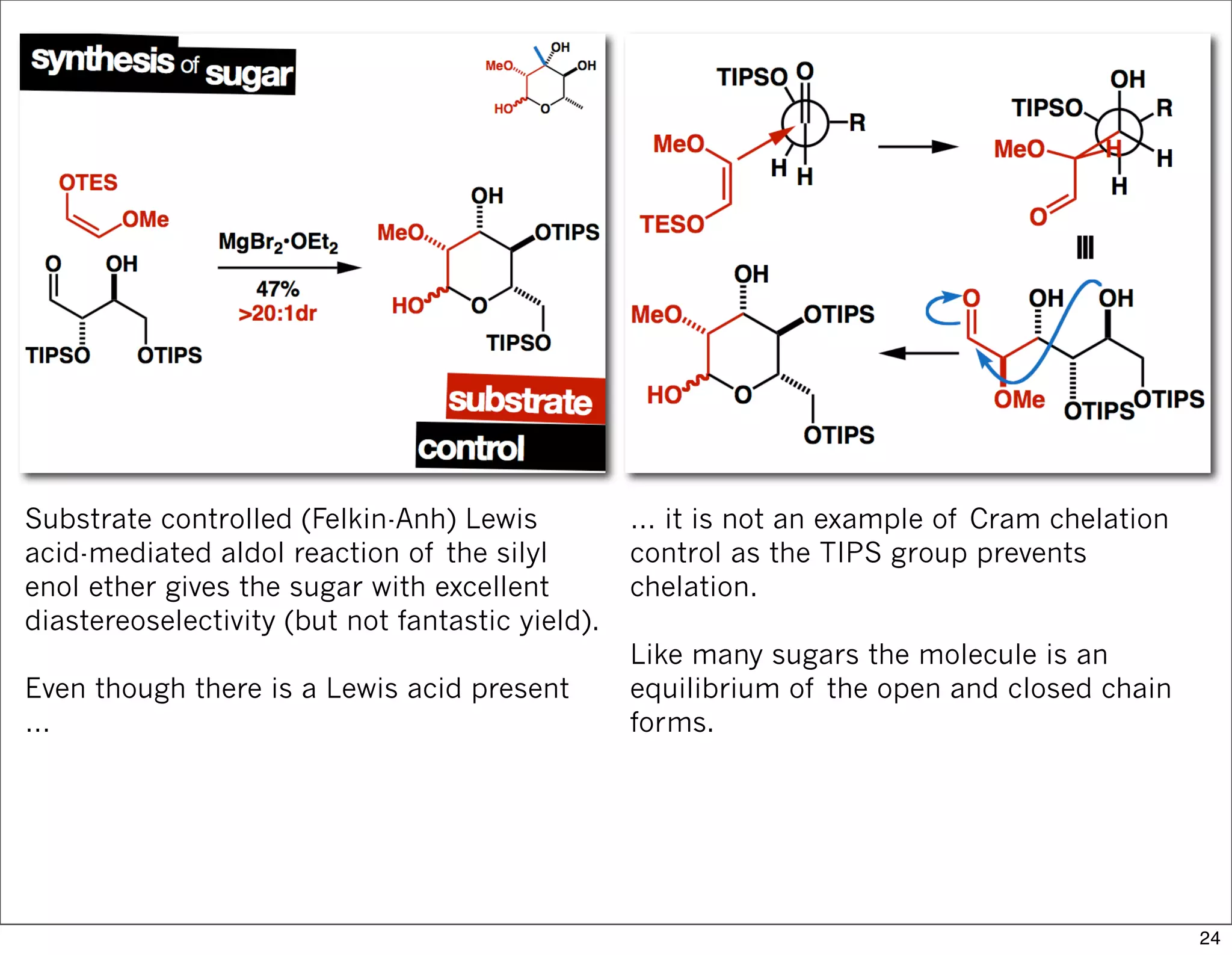
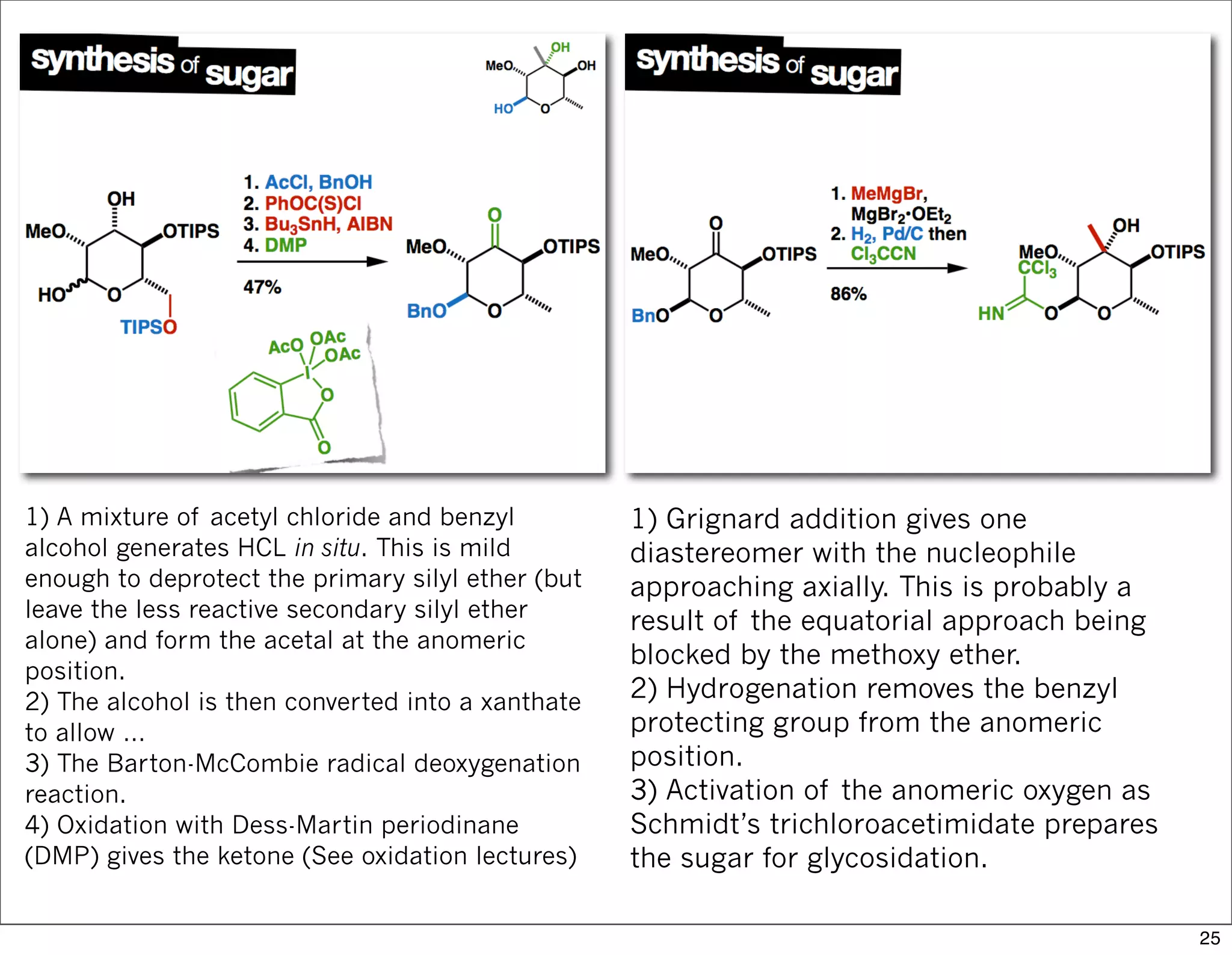
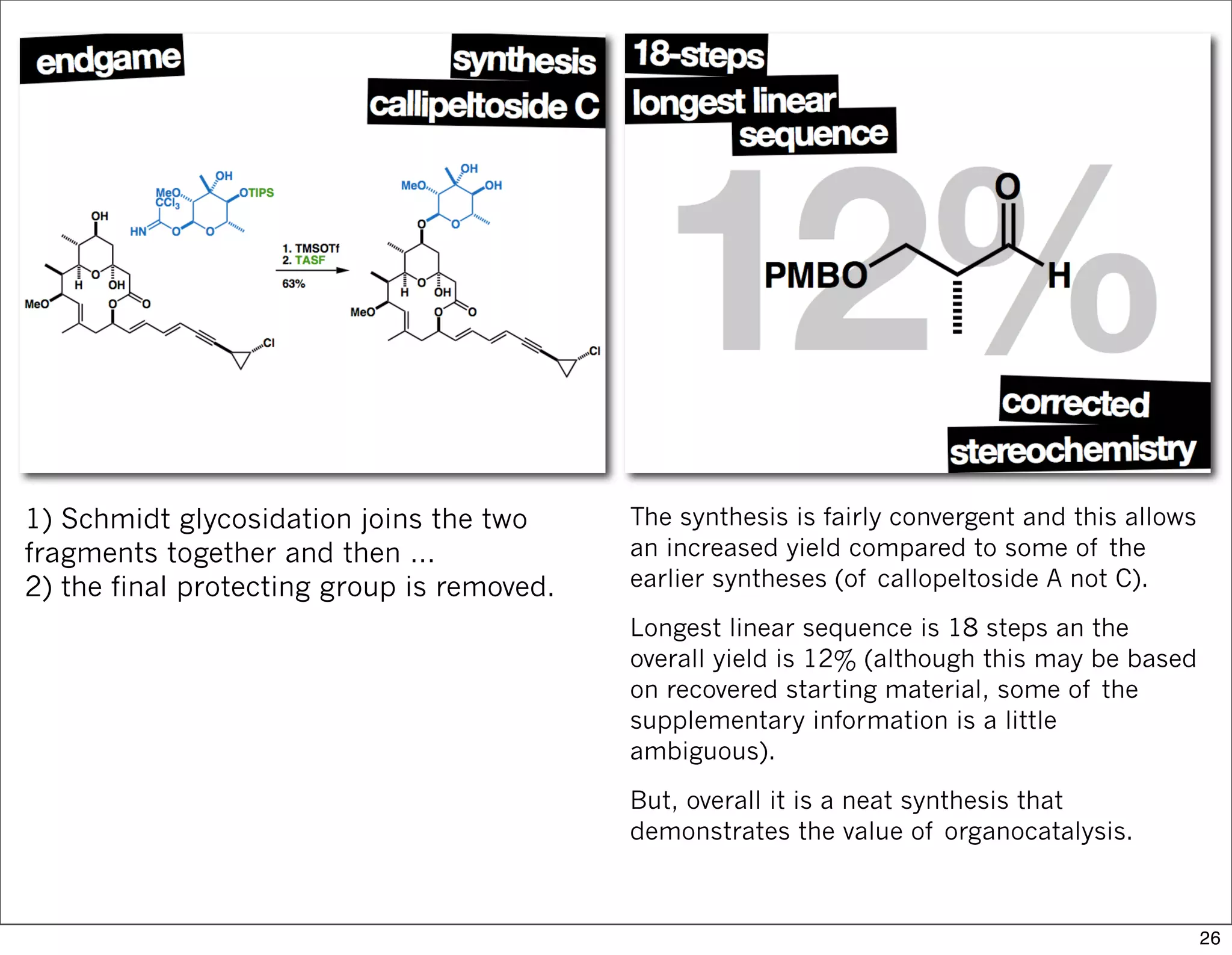

This document summarizes MacMillan's total synthesis of callipeltoside C, which employs organocatalysis and several interesting chemical transformations. The retrosynthesis splits the molecule into three fragments - the macrocyclic lactone core, carbohydrate, and a third segment prepared using organocatalysis. The forward synthesis couples these fragments in a convergent manner, with key steps including a Negishi carbometallation, organocatalytic hydroxylation, Semmelhack reaction to form the tetrahydropyran ring, and glycosidation to join the sugar moiety. The synthesis highlights the utility of retrosynthesis in simplifying complex targets and total synthesis in confirming the structure of natural products.
























































































