Downloaded 23 times
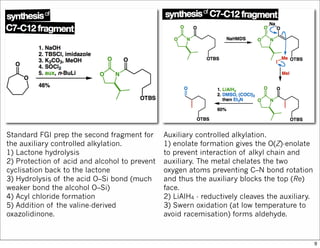
![1) Acetal formation - the reaction
conditions are sufficiently acidic to
promote deprotection of the primary silyl
ether (TBS = tert-butyldimethylsilyl).
2) Oxidative cleavage of the alkene. This
employs a substoichiometric quantity of
OsO4 and stoichiometric NaIO4. The OsO4
mediates dihydroxylation while the NaIO4
re-oxidises the osmium and cleaves the
diol. This is analogous to ozonolysis.
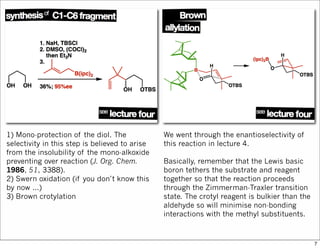
1) Addition of a Grignard reagent. We do
not care if the reaction is diastereoselective
or not because ...
2) Oxidises the alcohol to the ketone. TPAP
(tetrapropylammonium perruthenate [Pr4N]
[RuO4]) is a catalytic oxidant while NMO (N-
methylmorpholine N-oxide) is the
stoichiometric oxidant (it oxidises the
ruthenium).
That finishes the first fragment.
8](https://image.slidesharecdn.com/123713ablecture10-161118000039/85/123713AB-lecture10-8-320.jpg)
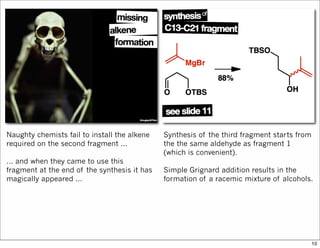
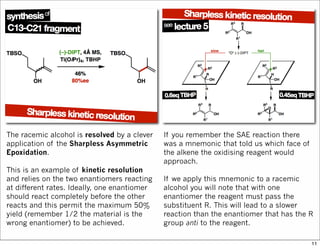
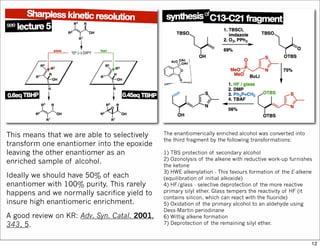

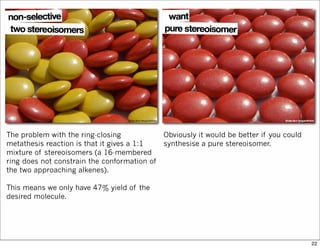
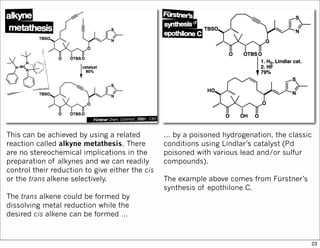
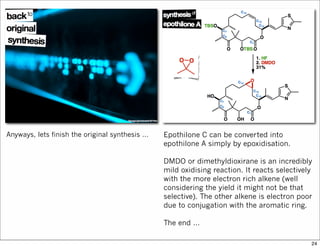

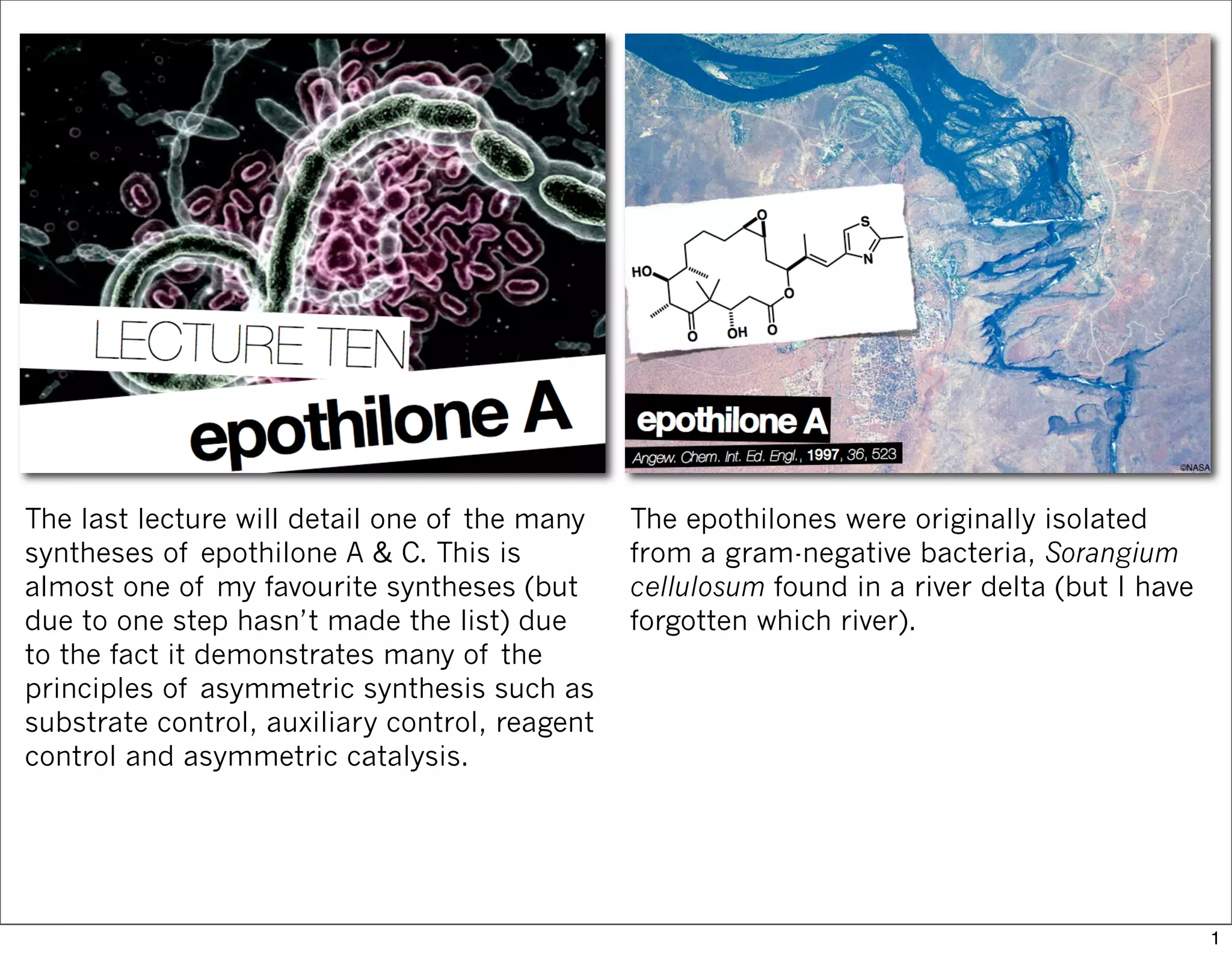
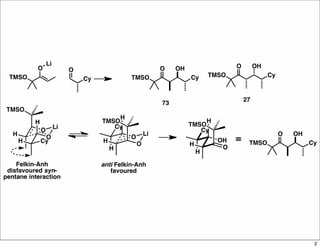
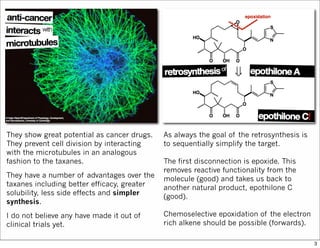
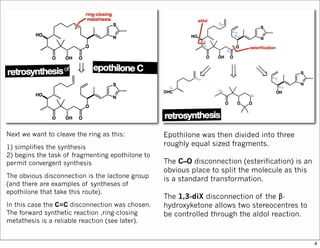
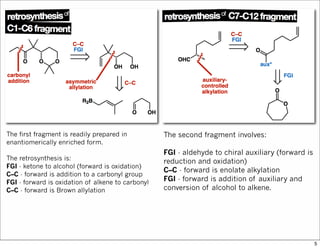
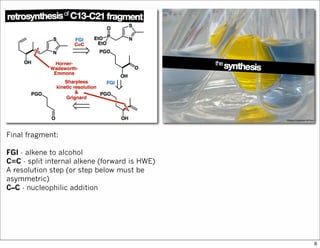
The document outlines a synthesis of epothilone A and C, emphasizing the principles of asymmetric synthesis and their potential as cancer drugs. It details the retrosynthetic analysis, fragmentation, and various synthetic steps involved, including reactive transformations and selective reactions. The concluding discussion highlights the use of ring-closing metathesis and the challenges related to stereochemistry in achieving desired yields.



































































