Downloaded 47 times

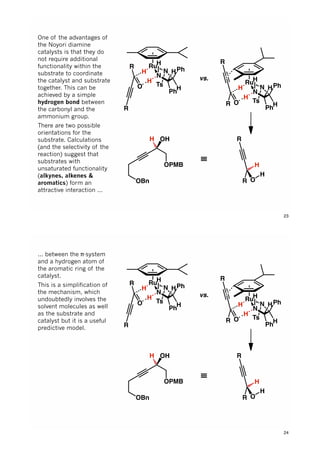
![PMBO
OHH
OBn
H
H
HO
O
DCC, DMAP
80%
PMBO
OBn
O
O
i. LiHMDS, TMSCl,
–78°C to rt
ii. CH2N2
79%
O
O
OBn
PMBO
DCC =
dicyclohexylcarbodiimide
DMAP =
dimethylaminopyridine
LiHMDS = lithium
hexamethyldisilazide
[lithium
bis(trimethylsilyl)amide]
Question 8
If you completed last weeks problems then this
sequence should offer no challenge (if you haven’t
looked at last weeks questions then you can either cheat
and look at the answers (thus spoiling the enjoyment) or
have a go now).
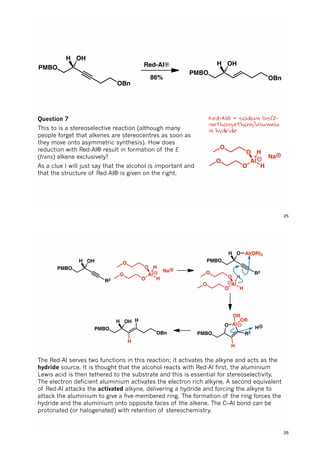
Reagents are given on the right.
N
C
N
N
N
27
PMBO
OBn
O
O
PMBO
OBn
O OTMS
Answer
The first step is esterification and I will not go through the mechanism of this reaction
(if you don’t know it or can’t work it out go through your undergraduate notes, it will in
be in there somewhere).
The second step involves stereoselective enolate/silyl ketene acetal formation. It is
essential that we control the geometry of the ‘enolate’ as this effects the relative
stereochemistry of the two new stereocentres formed in the rearrangement.
Under standard conditions such as this esters invariably give the (OSi)-E-enolate. One
simplistic interpretation of the Ireland model says this is due to the fact the ester
substituent can rotate out of the way so is smaller than the substituents of the base
and hence the enolate substituent will be better accommodated on the same face as
the alkoxy substituent.
28](https://image.slidesharecdn.com/8problems2answers-161117231324/85/Problems-2-answers-14-320.jpg)


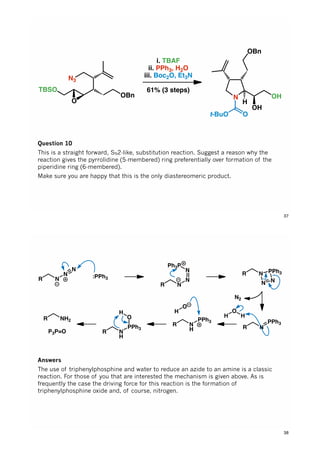


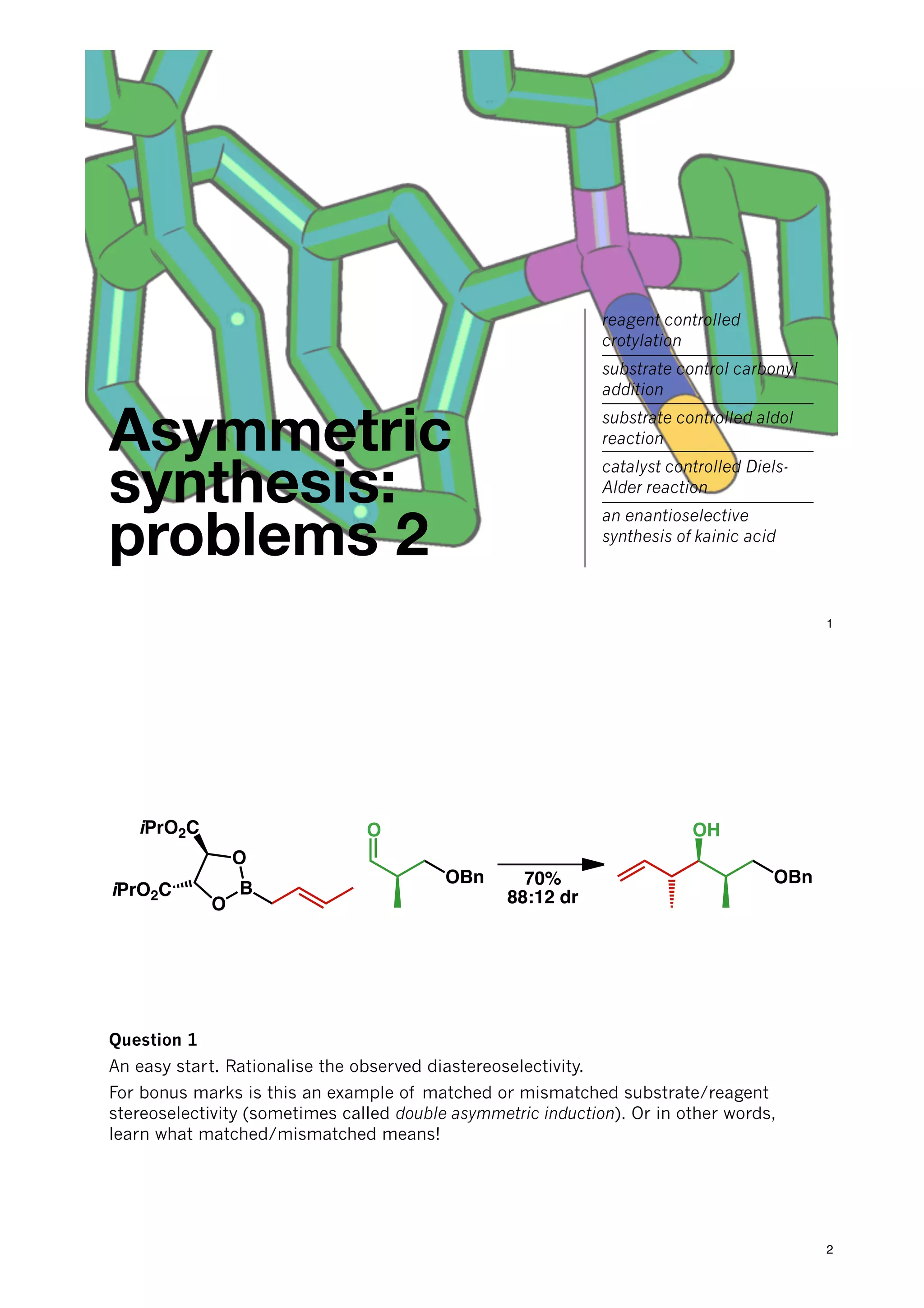
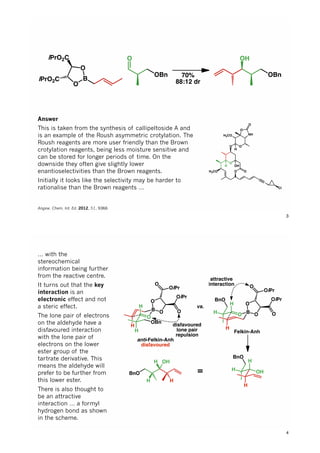
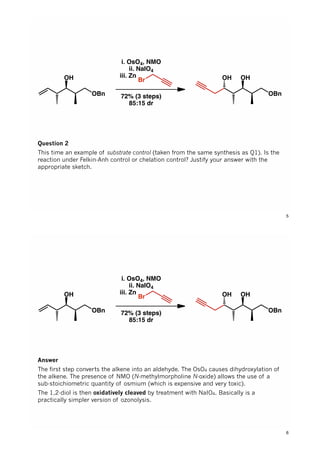
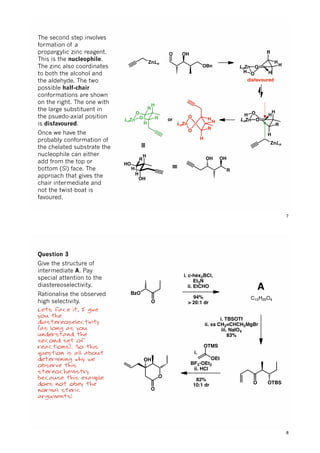
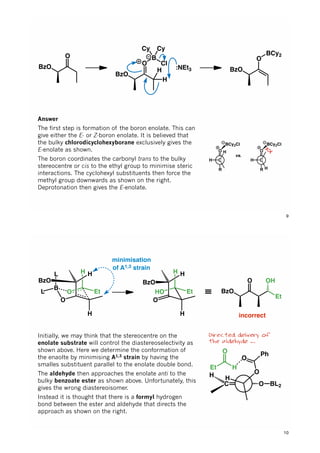
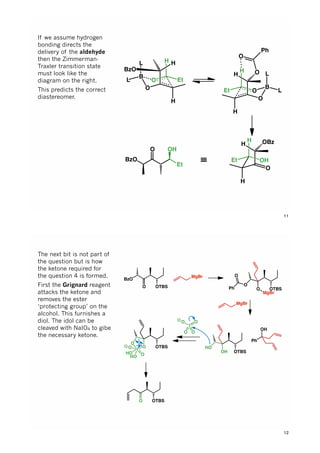
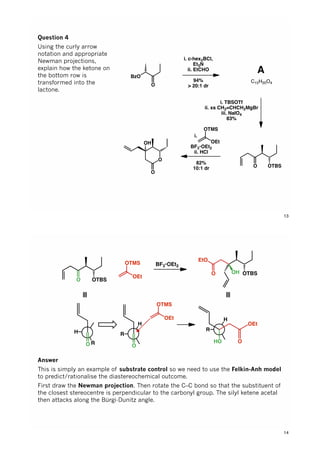
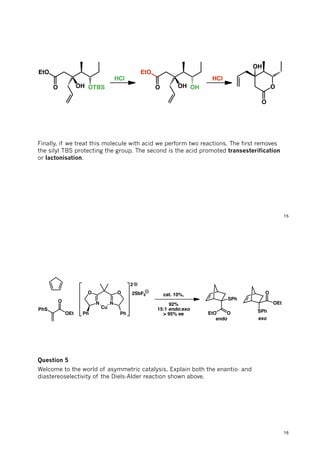
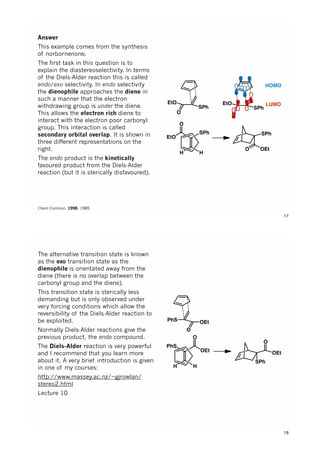
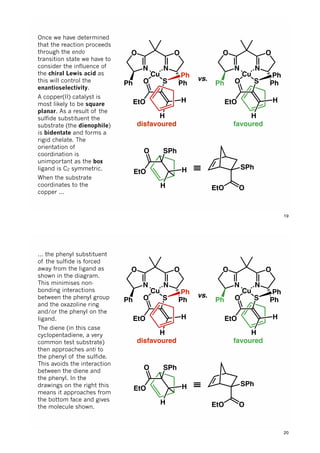
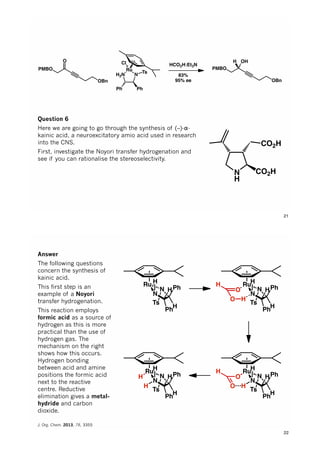
The document discusses various approaches in asymmetric synthesis, including crotylation, aldol reactions, and Diels-Alder reactions, focusing on selectivity and stereochemical outcomes. Key interactions in reactions, such as electronic and steric effects, are analyzed alongside specific synthesis examples of compounds like kainic acid. Each section addresses different synthesis steps, reacting conditions, and rationales for enantio- and diastereoselectivity in complex molecular reactions.