Download as PDF, PPTX


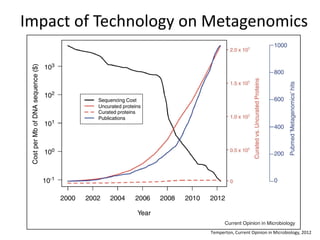
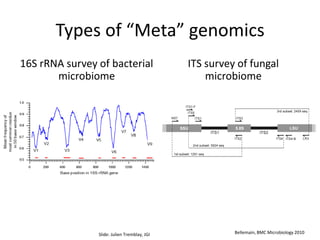








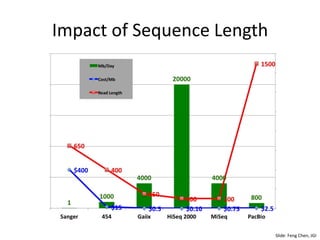
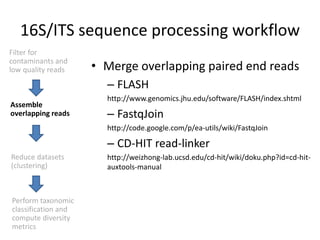
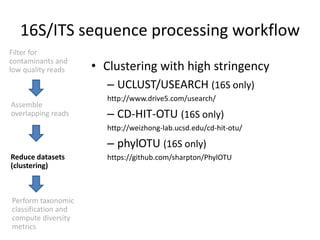



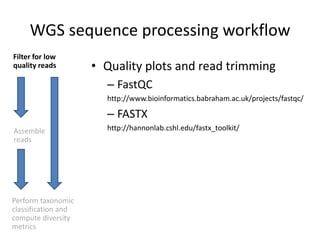
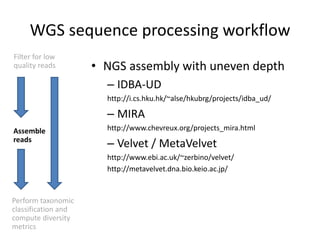
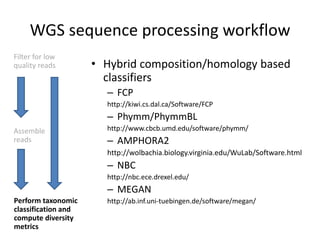
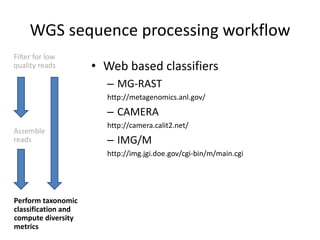


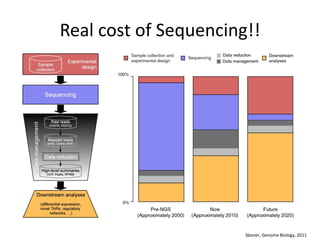



The document discusses computational tools for metagenomics, focusing on various types of metagenomic studies including 16S rRNA surveys and whole genome shotgun sequencing. It highlights the challenges and advantages of these methods, such as data processing workflows, amplification biases, and the need for high coverage in sequencing. Further, it emphasizes the importance of planning informatics workflows and incorporating statistical analyses early in the research process.











































































































