Download as PDF, PPTX














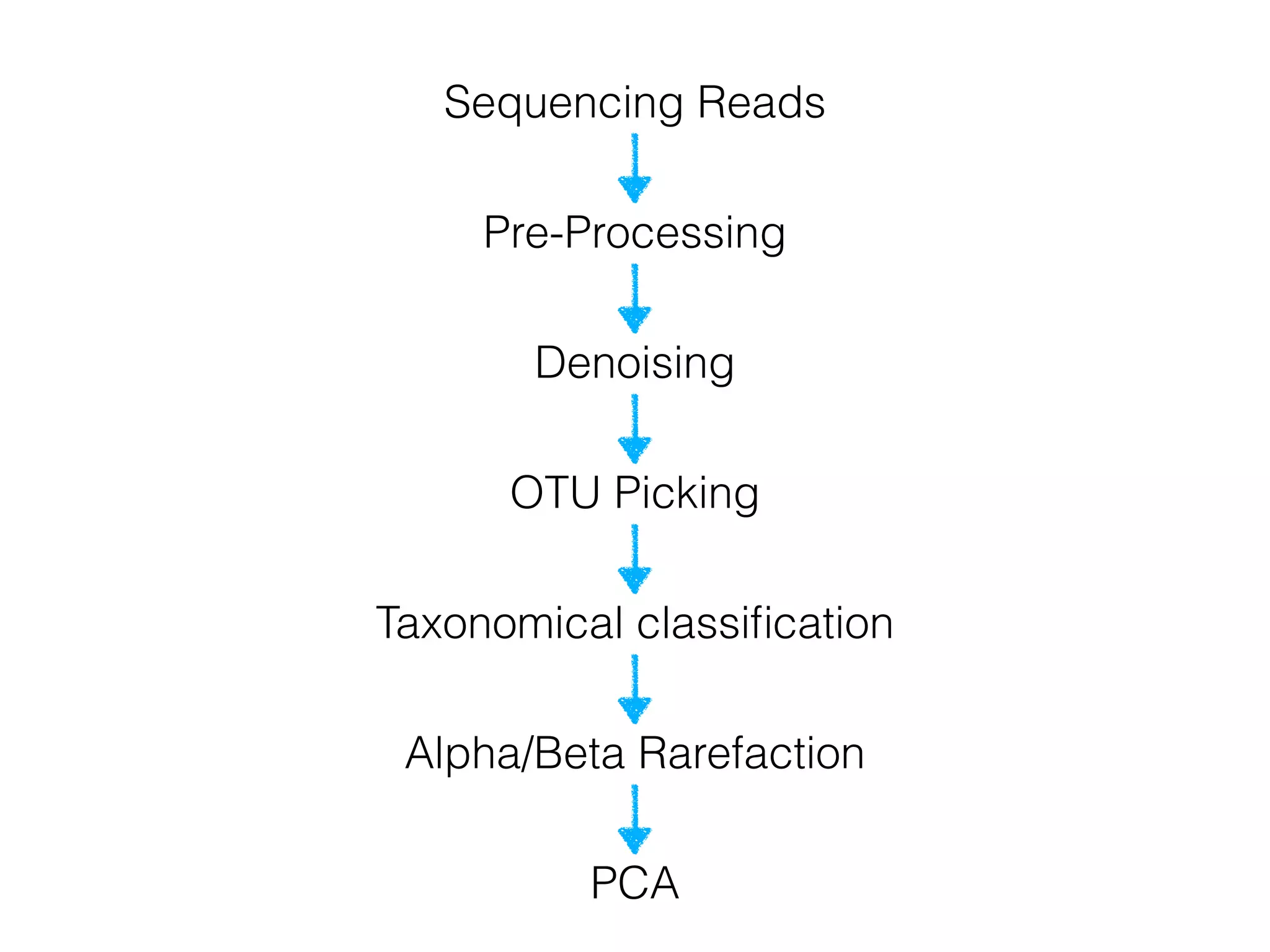
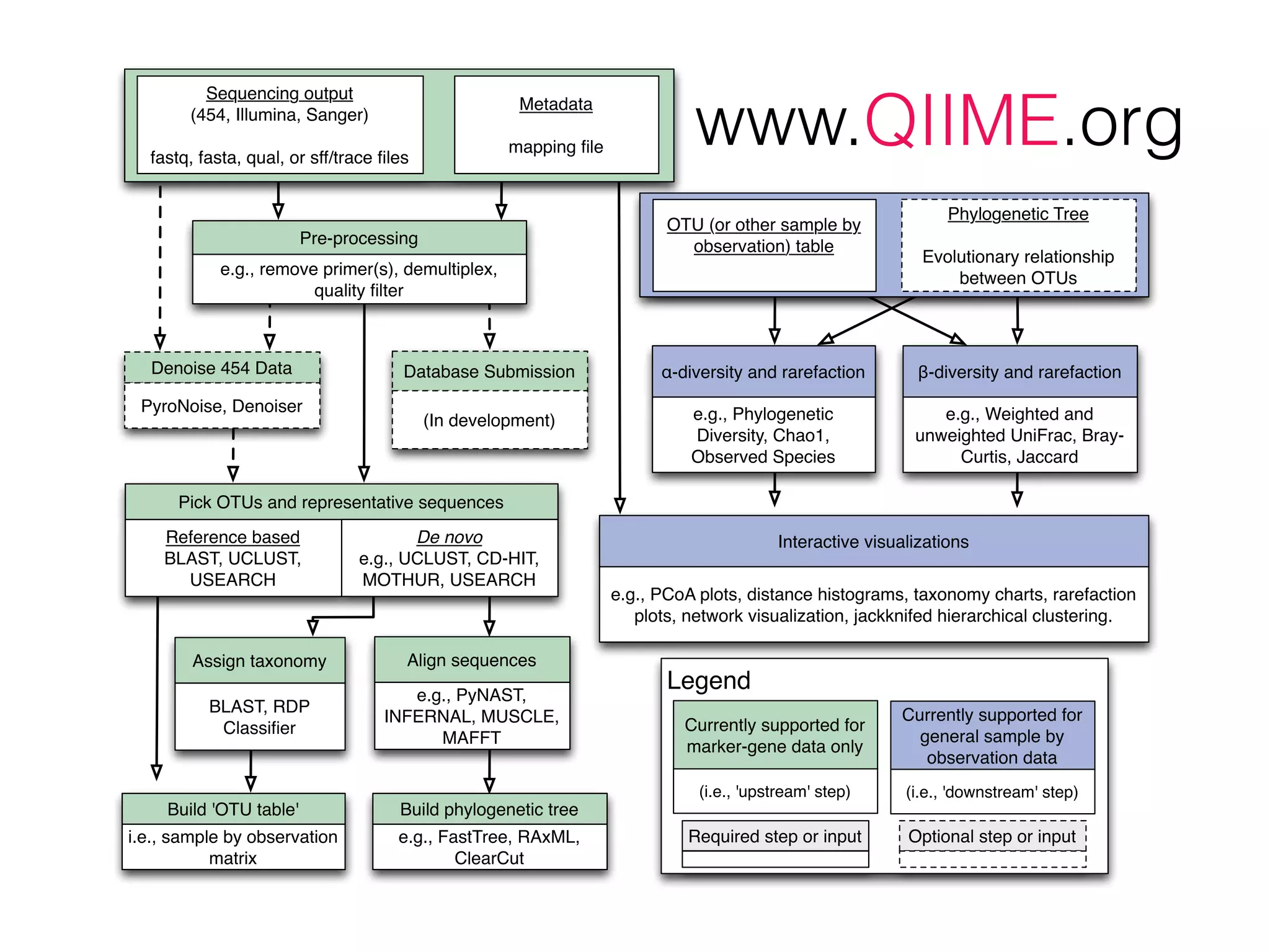
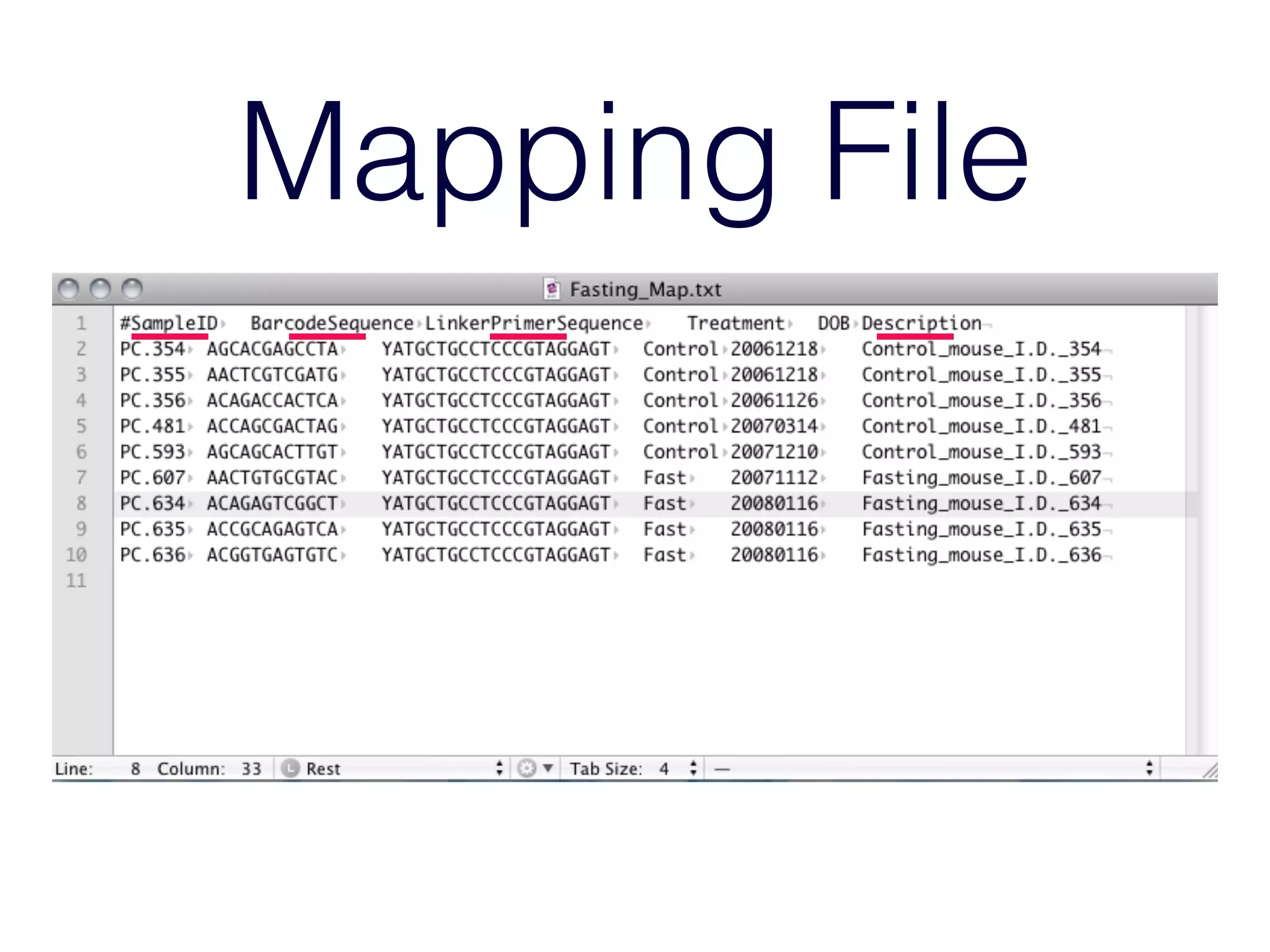






This document provides an overview and primer on 16S amplicon sequencing and analysis for metagenomics. It discusses how 16S is a ubiquitous gene that can be used to compare microbial communities across samples, outlines common analysis steps like preprocessing, OTU picking, taxonomy assignment, and diversity metrics, and introduces two analysis tools - MEGAN and Qiime. Key advantages and limitations of the 16S amplicon approach are highlighted.































![[13.09.19] 16S workshop introduction](https://cdn.slidesharecdn.com/ss_thumbnails/13-130923020358-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)


































































