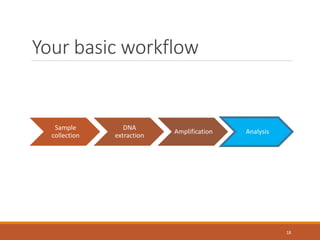
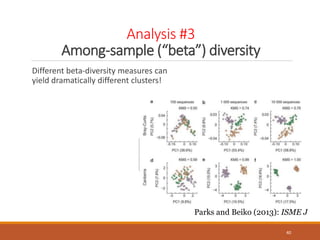
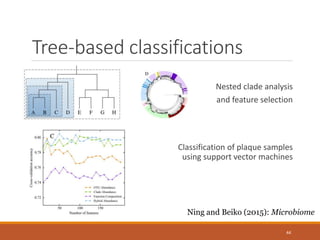
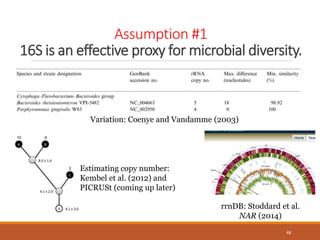
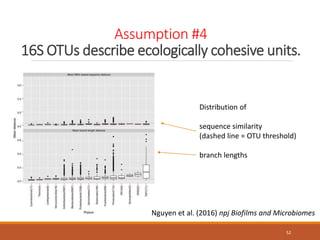
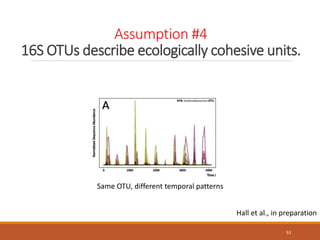
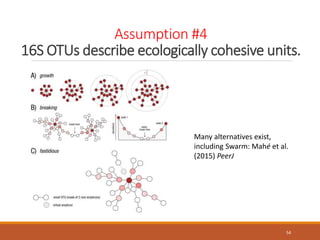
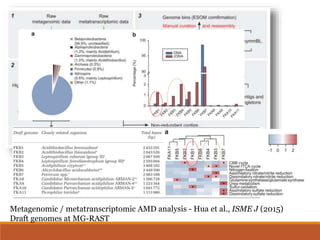
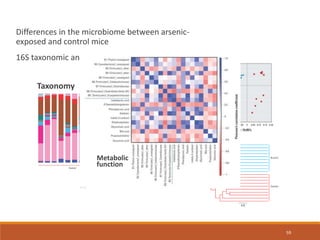
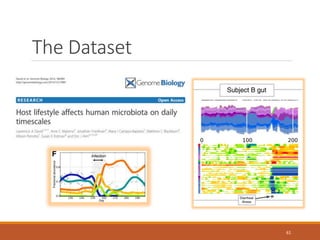
This document provides an overview of a tutorial on analyzing microbiome data using 16S rRNA gene sequencing and metagenomics. The morning session covers the basics of 16S analysis including sample collection, PCR amplification of the 16S gene, clustering sequences into OTUs, assigning taxonomy, and calculating alpha and beta diversity. The assumptions and limitations of 16S analysis are also discussed. The afternoon session introduces metagenomics and compares it to 16S analysis. It covers taxonomic and functional profiling from metagenomic data as well as tools like PICRUSt for predicting gene functions. The document concludes by discussing the value of multi-omics approaches that integrate different types of microbiome data.














![Sample collection and DNA extraction
Defined protocols exist, many kits (e.g. PowerSoil®)
Need to consider barriers to DNA recovery and PCR (e.g. humic acids
from soil, bile salts from feces)
Additional mechanical approaches (e.g., mechanical lysis of tissues with
bead beating)
Kits and rogue lab DNA can end up in your sample – need to run
negative controls!!
◦ Example from [year redacted]: shocking finding of bacterial DNA in the
[location redacted]! However, [taxonomic group redacted] was a known
frequent contaminant of DNA extraction kits.
19](https://image.slidesharecdn.com/ccbctutorialbeiko-160516183954/85/CCBC-tutorial-beiko-19-320.jpg)

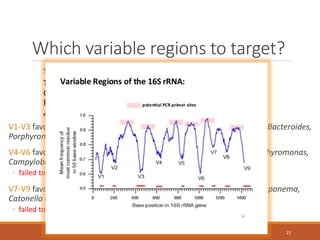
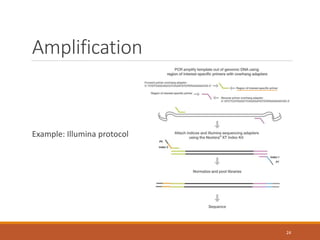
![Choosing a PCR strategy
Need to consider:
◦ Correct melting temperature (60-65 degrees C for Illumina
protocol)
◦ DNA sequencing read length (influences choice of primers)
◦ Primer specificity!
◦ Comparability with previous studies?
[Good luck with that]
[but that’s what the Earth Microbiome Project protocol
http://www.earthmicrobiome.org/emp-standard-protocols/16s/
is meant to achieve]
21](https://image.slidesharecdn.com/ccbctutorialbeiko-160516183954/85/CCBC-tutorial-beiko-21-320.jpg)





























































![[13.07.07] karst mewe13 dna_extraction_nonotes](https://cdn.slidesharecdn.com/ss_thumbnails/13-07-07karstmewe13dnaextractionnonotes-130707080129-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)






![[13.09.19] 16S workshop introduction](https://cdn.slidesharecdn.com/ss_thumbnails/13-130923020358-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)































































