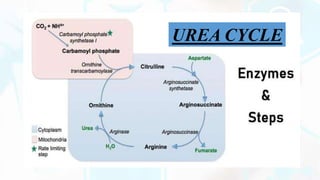
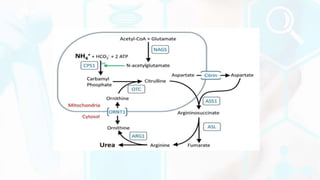
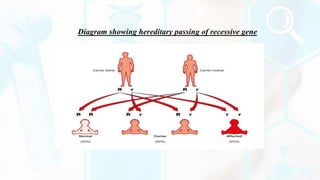
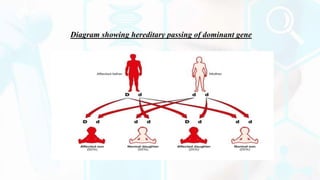
The document provides a detailed overview of the urea cycle, its enzymes, regulation, and associated disorders. It explains how ammonia, a toxic byproduct of amino acid breakdown, is converted into urea for safe excretion via the kidneys, highlighting the critical role of liver enzymes. Additionally, it discusses urea cycle disorders, their genetic basis, symptoms, and potential treatments, emphasizing the importance of timely intervention to prevent severe outcomes.