Download as PDF, PPTX















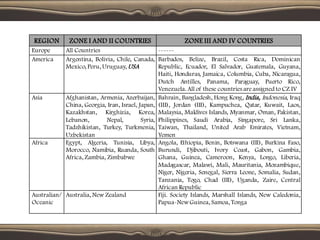



















![• Significant change occurs due to accelerated stability testing at an
intermediate condition [30°c +/- 2°c/ 60% RH +/- 5%] is defined as:
A 5% potency loss from the initial assay value of a batch.
Any specified degradant exceeding its specification limit.
The product exceeding its pH limits.
Dissolution exceeding the specification limit for 12 capsules/tablets.
Failure to meet specifications for appearance and physical properties.](https://image.slidesharecdn.com/stability-240325124850-227b13d2/85/STABILITY-STUDIES-OF-PHARMACEUTICALS-pdf-35-320.jpg)









The document provides guidelines for stability testing of pharmaceuticals. It defines stability testing as tests designed to obtain information on a product's stability to define its shelf life and utilization period under specified storage conditions. The objectives are to select adequate formulations, determine shelf life and storage conditions, substantiate the claimed shelf life, and verify no changes adversely affecting stability. The guidelines address information required for new drug substances and products and covers phases of testing including development, registration, and post-registration. It provides details on design of studies for drug substances and products, storage conditions, testing frequency and evaluation.























































































