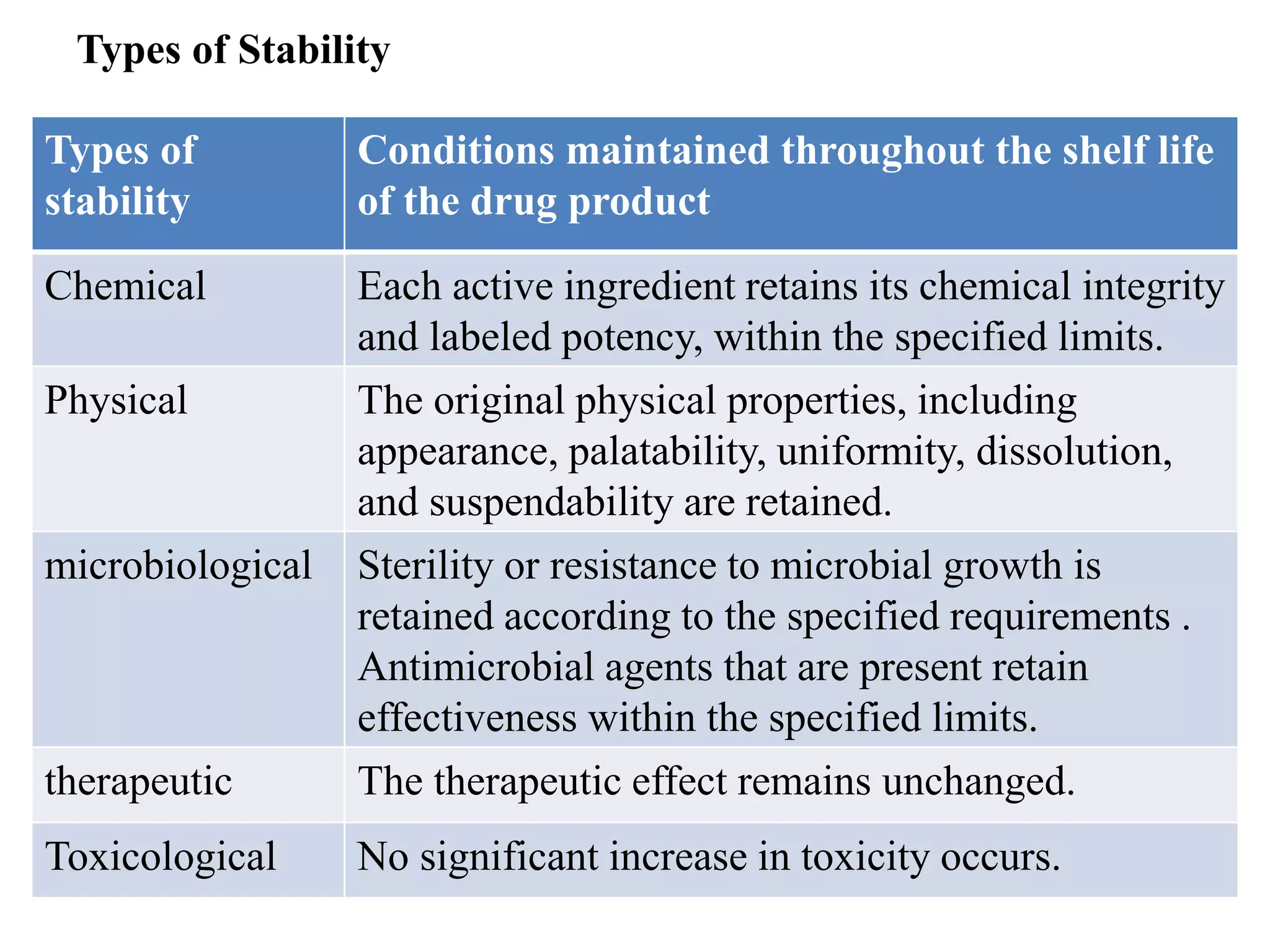
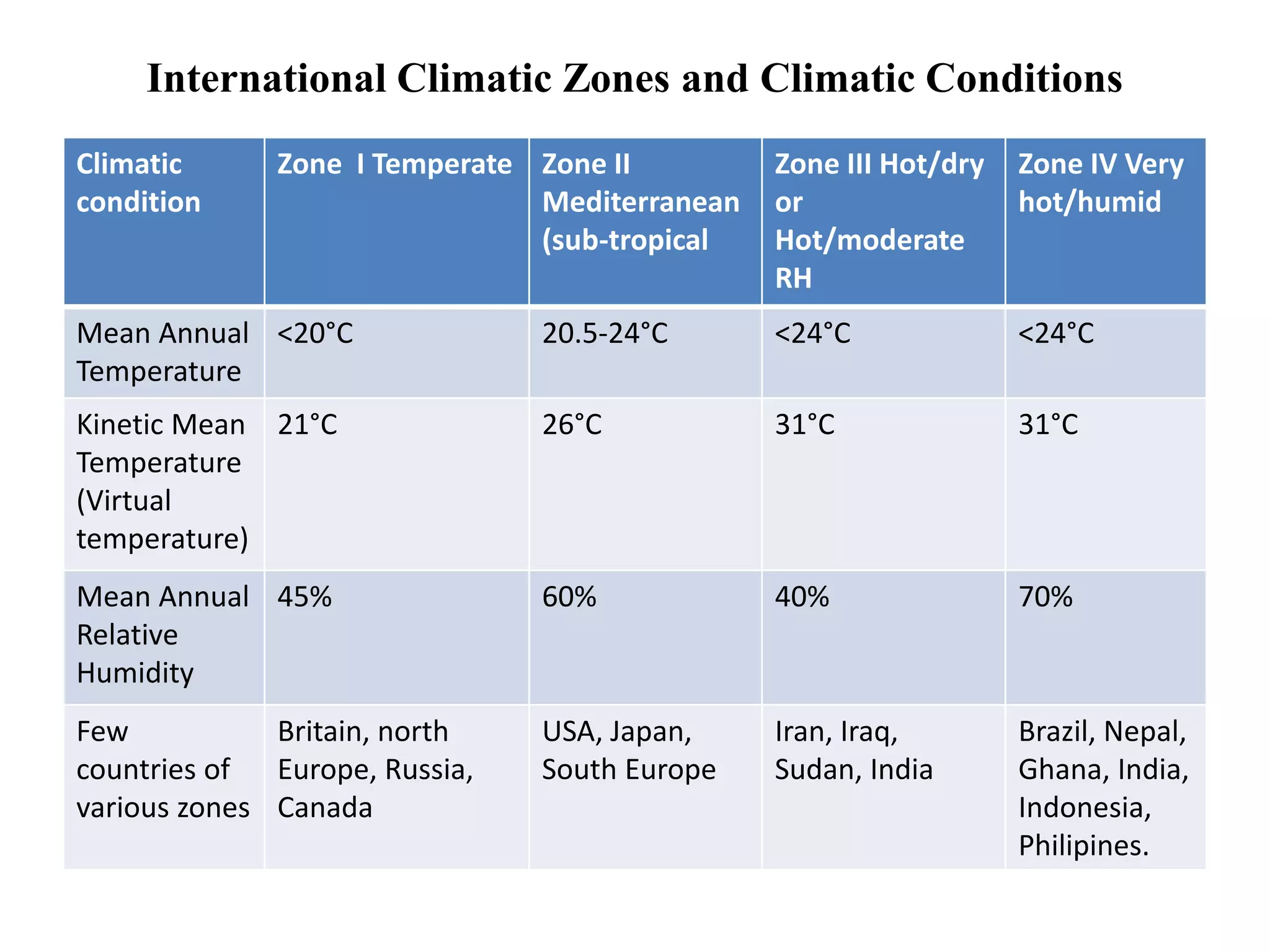
The document discusses the purpose and types of stability studies for pharmaceutical products. It defines stability as the capability of a formulation to remain within specifications for identity, purity, quality and strength throughout its defined shelf life under recommended storage conditions. The main types of stability are chemical, physical, microbiological, therapeutic and toxicological. Stability testing aims to provide evidence of how quality varies over time under environmental factors like temperature, humidity and light. It also addresses product-related factors like interactions. Protocols, results and conclusions from stability studies should be summarized in the dossier. Ongoing stability programs are required to monitor products throughout their shelf life.

























































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)






