Downloaded 148 times



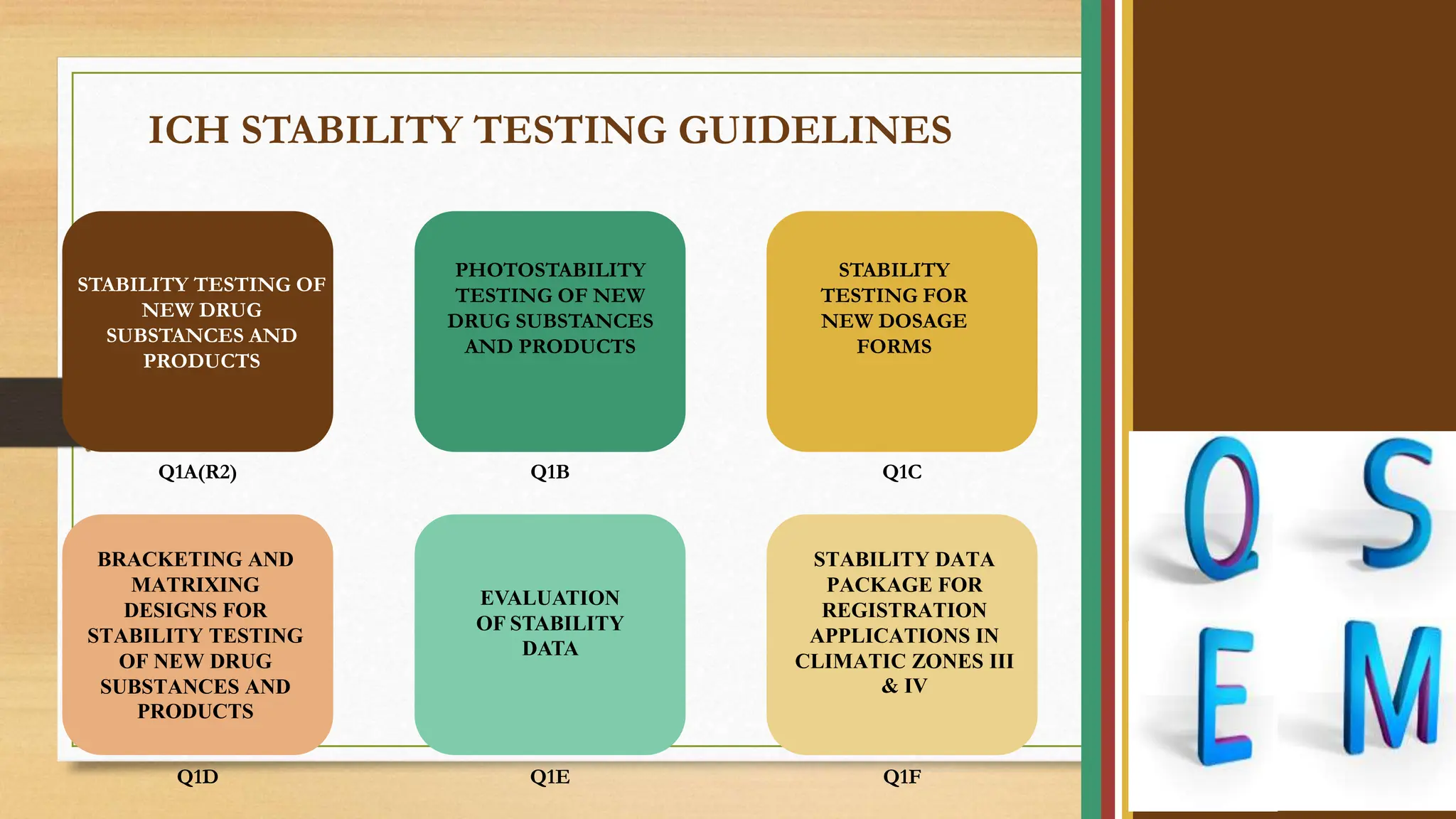

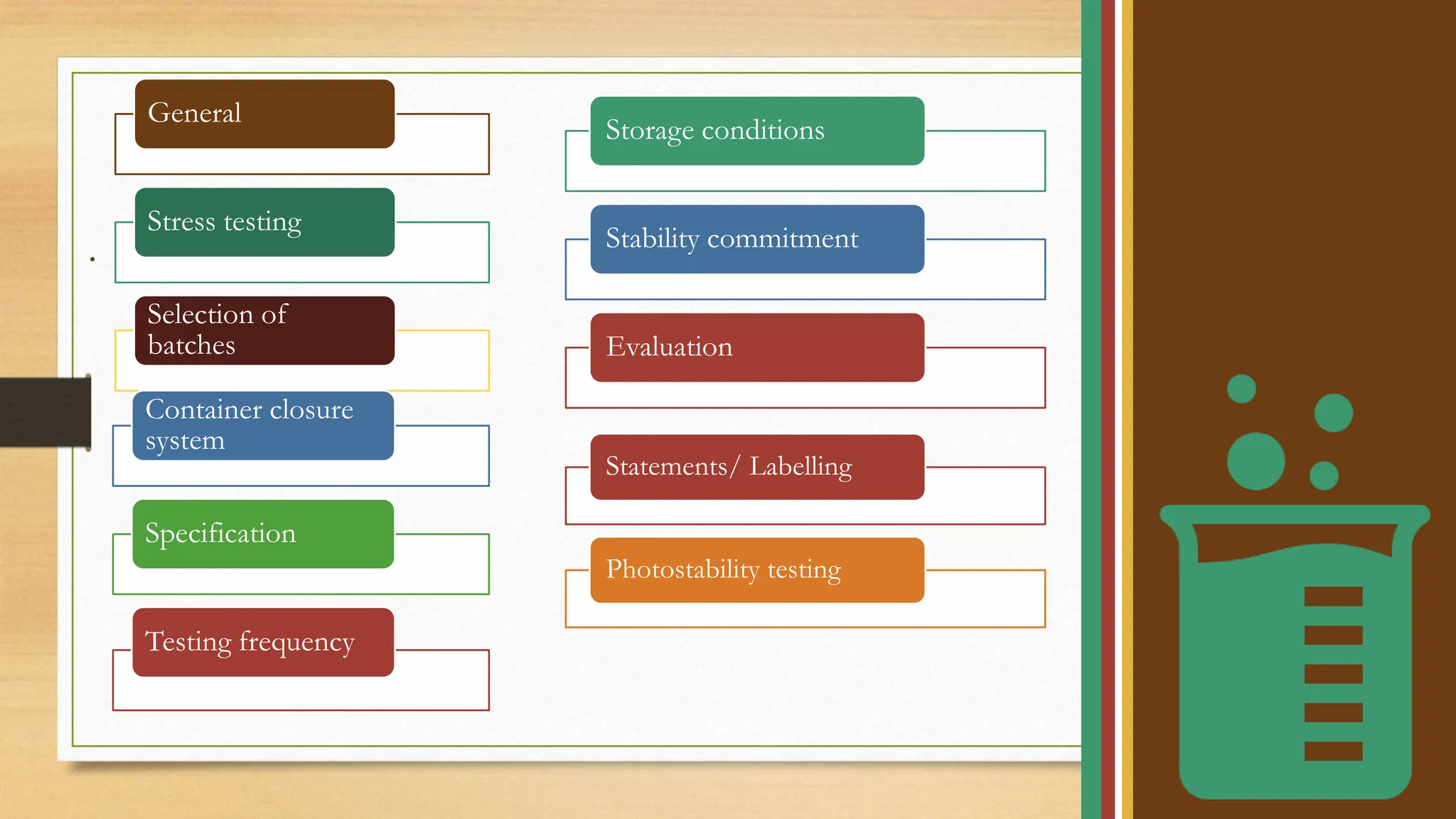



























The document outlines ICH stability testing guidelines for new drug substances and products, detailing environmental factors affecting drug quality over time. It includes protocols for stability testing, photostability, bracketing and matrixing designs, as well as recommendations for evaluating stability data, particularly in different climatic zones. The aim is to ensure the safety, quality, and efficacy of pharmaceuticals through systematic testing and data evaluation.

















































































