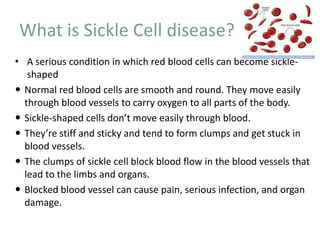
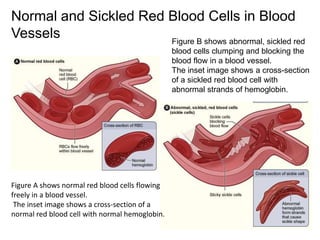
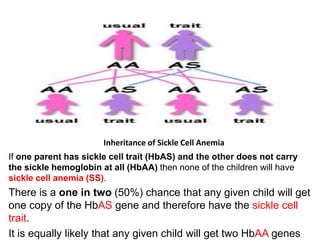
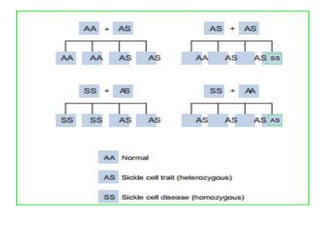
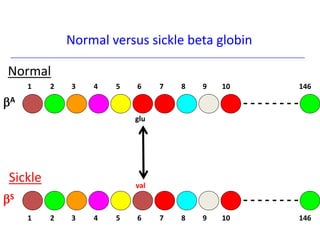
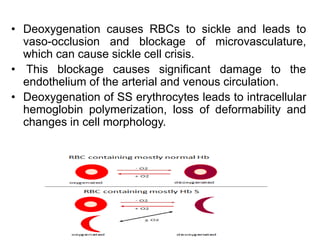
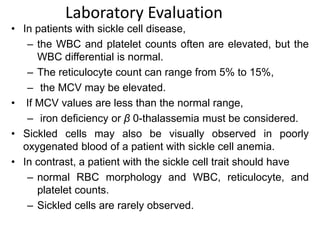
The document discusses sickle cell disease, describing how a genetic mutation causes red blood cells to become sickle-shaped and get stuck in blood vessels, blocking blood flow and oxygen delivery. Treatment aims to reduce symptoms, complications, and crises through medications like hydroxyurea and penicillin prophylaxis, transfusions, pain management, and lifestyle changes. New treatments under investigation include gene therapy, bone marrow transplants, and other drugs targeting hemoglobin levels and cell adhesion.