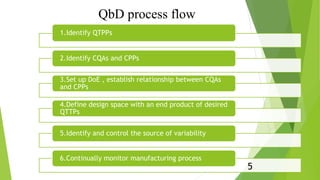
The document summarizes ICH Q8 guidelines for pharmaceutical product development using a Quality by Design (QbD) approach. It discusses key QbD concepts like quality target product profiles, critical quality attributes, critical process parameters, and design space. The guidelines suggest determining aspects of drug substances, excipients, manufacturing processes, and container closure systems that are critical to quality. They provide guidance on contents for drug product development documentation, including formulation development, compatibility studies, container closure selection, and ensuring microbiological attributes and stability. The QbD approach aims to build quality into pharmaceutical products from the design stage through understanding and control of material and process variables.









































![Pulmonary drug delivery system [PDDS]](https://cdn.slidesharecdn.com/ss_thumbnails/pulmonarydrugdeliverysystem-160619063517-thumbnail.jpg?width=640&height=640&fit=bounds)








































