Downloaded 298 times




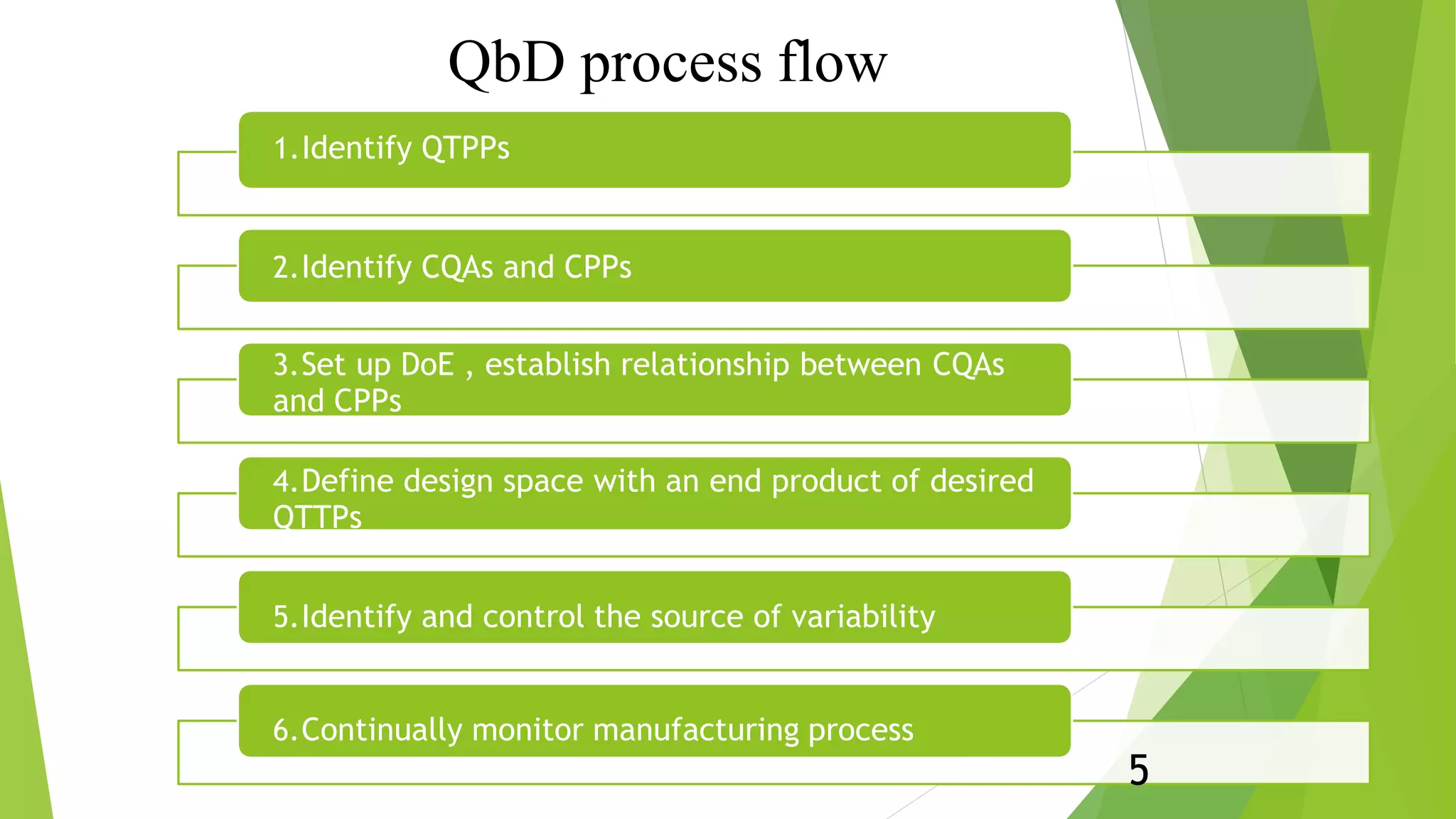











This document presents an overview of ICH Q8 guidelines for pharmaceutical product development using Quality by Design (QbD) principles. It discusses key QbD concepts like Quality Target Product Profile, critical quality attributes, critical process parameters, and design space. The document also summarizes the contents that should be included in the CTD quality module regarding drug substances, formulation development, manufacturing process, container closure system, microbiological attributes, and compatibility studies. Finally, it emphasizes that QbD ensures quality is built into the product design rather than relying solely on end-product testing.























































































