Download as PDF, PPTX

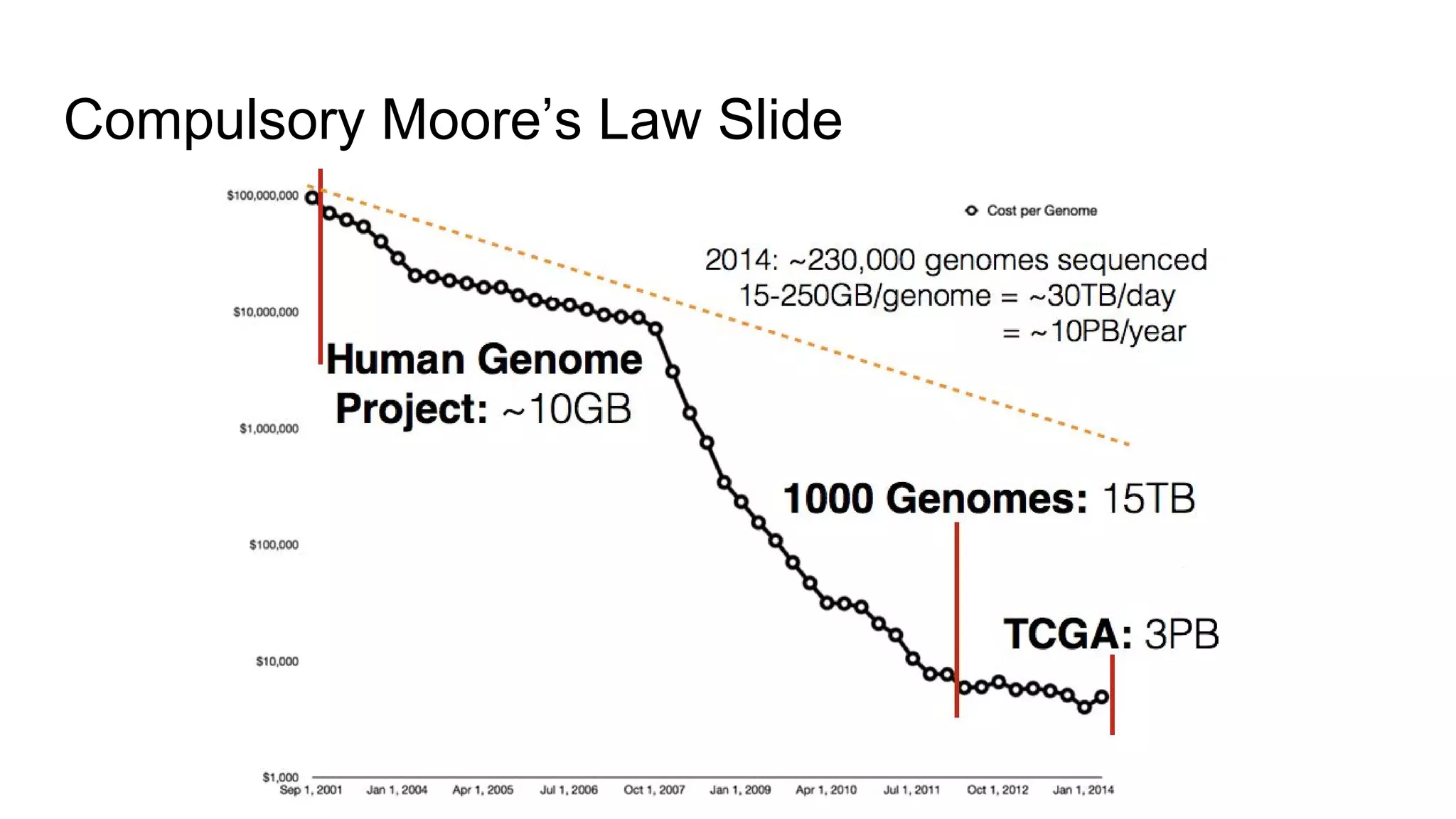





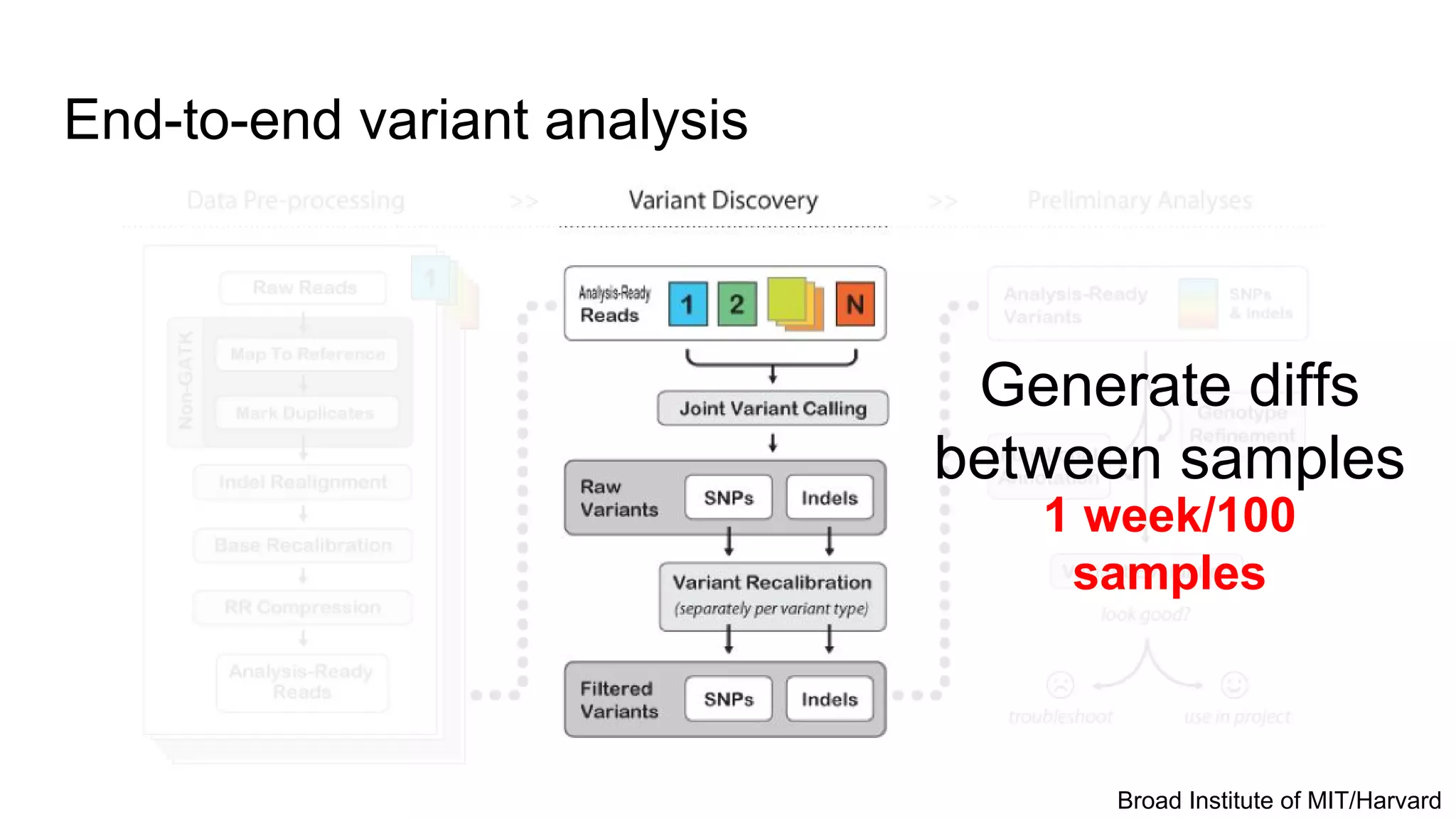
















This document discusses analyzing large genomic datasets with ADAM and Toil. It summarizes the sequencing and analysis process, and how ADAM implemented on Spark can provide horizontal scalability and speedups of 30-50x over traditional tools. Toil is introduced as a pipeline system for massive genomic workflows that can run on thousands of nodes and is resilient to failures. Results show ADAM produces equivalent variants to GATK while being 3.5x faster and 4x cheaper.











































































































